Location: Home >> Detail
Crop Breed Genet Genom. 2026;8(1):e260002. https://doi.org/10.20900/cbgg20260002
 ,
Mbe Joseph Okpani ,
Ihuoma Umezurumba Okwuonu ,
Chiedozie Ngozi Egesi
,
Mbe Joseph Okpani ,
Ihuoma Umezurumba Okwuonu ,
Chiedozie Ngozi Egesi
National Root Crops Research Institute, Umudike, Umuahia P.M.B. 7006, Abia State, Nigeria
* Correspondence: Simon Peter Abah
Cassava (Manihot esculenta Crantz) is a critical tropical root crop that underpins food security, industrial applications, and rural livelihoods, particularly across sub-Saharan Africa. Traditional cassava breeding has faced significant challenges due to the crop’s long growth cycle, clonal propagation, high heterozygosity, polygenic inheritance of key traits, and strong genotype × environment (G × E) interactions. These factors have historically limited the rate of genetic improvement, despite cassava’s socio-economic importance. Recent developments in genomics-assisted breeding (GAB) are transforming cassava improvement by combining high-density molecular markers, genome-wide association studies (GWAS), genomic selection (GS), and multi-omics approaches such as transcriptomics, metabolomics, and high-throughput phenotyping. The sequencing of the cassava reference genome has enabled single nucleotide polymorphism (SNP) discovery, quantitative trait loci (QTL) mapping, and the development of genomic prediction models, allowing early selection for complex traits including root yield, dry matter content, starch quality, carotenoid accumulation, disease resistance, and early storage root bulking. Functional genomics, together with non-invasive root imaging technologies such as ground-penetrating radar, provides insight into the genetic and physiological mechanisms underlying root development and stress responses. Multi-environment GWAS and genomic selection models now account for G × E interactions, improving prediction accuracy and supporting rapid-cycle breeding. Furthermore, integrating metabolomic and transcriptomic datasets enhances trait prediction, accelerates candidate gene discovery, and advances precision breeding strategies. This review provides recent progress, tools, and future prospects in genomics-assisted cassava breeding, illustrating how modern genomic and phenomic technologies are reshaping breeding pipelines, shortening breeding cycles, and facilitating the development of climate-resilient, high-yielding, and farmer-preferred cassava varieties.
Cassava (Manihot esculenta Crantz) is a staple food and industrial crop of global importance, providing dietary energy for over 800 million people and supporting rural livelihoods across sub-Saharan Africa, Latin America, and Southeast Asia [1,2]. Its ability to produce reasonable yields under marginal soils, intermittent drought, and low external inputs has made cassava a strategic crop for food security in the face of climate variability [3,4]. In Africa, particularly Nigeria, the world’s largest producer, cassava underpins household food systems, starch-based industries, and emerging bioeconomy value chains [5,6].
Despite its resilience and socio-economic relevance, genetic improvement in cassava has lagged behind that of major cereal crops. This slow progress is largely attributable to inherent biological and genetic constraints, including a long breeding cycle (8–18 months), clonal propagation, high heterozygosity, and severe inbreeding depression [7–9]. Moreover, most agronomically and industrially important traits in cassava, such as storage root yield, dry matter content, starch quality, carotenoid accumulation, and cyanogenic potential, are quantitatively inherited and strongly influenced by genotype × environment (G × E) interactions [10,11]. These features reduce selection efficiency under conventional phenotypic breeding and necessitate long, resource-intensive multi-location testing schemes [12].
Over the past two decades, cassava breeding has undergone a major transformation driven by advances in genomics, particularly the release of high-quality reference genome assemblies and the development of cost-effective, high-density single nucleotide polymorphism (SNP) genotyping platforms [13–15]. These genomic resources, combined with declining genotyping costs, have enabled the systematic application of genomics-assisted breeding (GAB) approaches in cassava, including marker-assisted selection (MAS), genomic selection (GS), genome-wide association studies (GWAS), and more recently, genome editing using CRISPR/Cas systems [11,16–19].
Marker-assisted selection has been successfully deployed in cassava breeding programs, particularly for traits controlled by major-effect loci such as resistance to cassava mosaic disease (CMD) and cassava brown streak disease (CBSD) [9,11,16,17,20]. However, its effectiveness diminishes for complex quantitative traits, including storage root yield, dry matter content, and starch quality, which are governed by many small-effect loci and are highly influenced by genotype × environment interactions [10,11]. These limitations have motivated a shift toward genomic selection, which leverages genome-wide marker information to capture cumulative additive genetic effects without requiring prior knowledge of individual quantitative trait loci [21,22].
Empirical studies have demonstrated the potential of genomic selection to improve prediction accuracy and reduce breeding cycle length in cassava, although reported accuracies vary widely across traits, populations, and environments [23]. Variability in GS performance has been attributed to differences in training population size, genetic relatedness between training and prediction sets, marker density, phenotyping precision, and environmental heterogeneity [22,24]. These findings highlight the critical role of high-quality phenotyping and multi-environment data in realizing the full benefits of genomic prediction in cassava breeding.
In parallel, GWAS has contributed to the identification of genomic regions and candidate genes associated with key agronomic and quality traits, including root architecture, carotenoid accumulation, cyanogenic potential, and disease resistance [16,17,25,26]. While GWAS has enhanced biological understanding of trait architecture, the translation of association signals into routine breeding decisions remains challenging due to small effect sizes and limited reproducibility across environments.
More recently, genome editing technologies, particularly CRISPR/Cas-based systems, have emerged as powerful tools for functional genomics and targeted trait improvement in cassava. Successful edits have been reported for genes involved in disease resistance, starch biosynthesis, and cyanogenic glucoside metabolism [27,28]. Nevertheless, the widespread adoption of genome editing in cassava breeding is constrained by low transformation efficiency, genotype dependency, regulatory uncertainty in many cassava-growing regions, and incomplete functional annotation of the cassava genome [19,29].
This review is therefore guided by the central hypothesis that integrating genomic tools with high-quality phenotyping and environmental information can substantially enhance selection accuracy and accelerate genetic gain in cassava breeding, particularly for complex, polygenic traits. By examining empirical evidence across MAS, GS, GWAS, and genome editing studies, this review evaluates how genomics-assisted breeding has reshaped cassava improvement efforts, identifies critical bottlenecks limiting its full adoption, and highlights emerging opportunities for next-generation breeding frameworks.
Genomic-Assisted Breeding as a Catalyst for Accelerated ImprovementThe advent of molecular markers, cost-effective sequencing, and high-resolution phenotyping has fundamentally transformed cassava improvement over the past decade. Genomics-assisted breeding (GAB) integrates next-generation sequencing (NGS), genome-wide association studies (GWAS), genomic selection (GS), and multi-omics data (transcriptomics, metabolomics, and phenomics) to unravel trait architecture and accelerate breeding progress [11,30,31]. These innovations follow the sequencing of the cassava reference genome [32], which laid the foundation for large-scale SNP discovery, high-density SNP arrays, and genotyping-by-sequencing platforms [33].
Genomic prediction tools developed through international consortia, such as the NextGen Cassava Breeding Project, enable breeders to estimate genomic estimated breeding values (GEBVs) early in the selection cycle, bypassing lengthy field evaluations and enhancing response to selection [11,34]. Combined with high-throughput phenotyping technologies, including unmanned aerial systems, imaging platforms, and non-invasive root imaging methods such as ground penetrating radar [35], genomic tools allow quantification of complex traits that previously confounded traditional breeding.
Cassava (Manihot esculenta Crantz) exhibits a predominantly outcrossing reproductive system, a characteristic that has profound implications for its genetic structure and breeding behavior. Although a small degree of natural selfing does occur, estimated at 5%–10%, outcrossing is maintained largely through asynchronous flowering, spatial arrangement of male and female flowers on the same inflorescence, and innate self-incompatibility tendencies [9,36,37].
From a population genetics perspective, this mating system contributes to:
Extreme heterozygosity which means cassava cultivars carry numerous heterozygous loci accumulated through generations of vegetative propagation and outcrossing. This maintains broad genetic diversity within clones but complicates allele fixation.
High segregation in progeny which means sexual recombination results in marked phenotypic variability, even among full-sib families, often producing many inferior segregants and only a few elite recombinants [38].
Severe inbreeding depression which means attempts to self-pollinate cassava drastically reduces vigor, fertility, root yield, and even survival rates. This is attributed to the expression of deleterious recessive alleles that accumulate over clonal generations [39].
Difficulty generating homozygous lines, which means traditional line development, as practiced in cereals, is nearly impossible in cassava. The few cycles of selfing achievable often yield weak plants, thereby slowing the development of inbred lines needed for hybrid breeding [36].
These biological limitations fundamentally shape cassava breeding strategies, forcing reliance on clonal selection, recurrent phenotypic evaluation, and more recently, genomic tools capable of capturing additive and non-additive genetic variance in highly heterozygous populations.
Storage Root FormationCassava storage root development is a complex physiological process integrating anatomical differentiation, secondary growth, carbon assimilation, and hormonal regulation. Unlike tuber crops such as potato, cassava storage roots originate from fibrous roots that undergo secondary thickening. Key developmental processes include:
Phloem parenchyma cell differentiation which involves the enlargement and starch deposition that occur primarily in the secondary phloem. The early differentiation of parenchyma cells sets the foundation for bulking capacity [40].
Secondary growth which involves the cambial activity, producing secondary xylem and secondary phloem, that drives root thickening. Varieties differ widely in how early and vigorously these tissues proliferate [41].
Carbohydrate partitioning which involves the allocation of photosynthates between shoots biomass and root sinks that determines early bulking. Genotypes with stronger sink strength and higher assimilate translocation tend to bulk earlier [42].
Hormonal control which involves the auxins that regulate cambial activity and root thickening, cytokinins promote cell division, abscisic acid (ABA) is associated with stress-induced root growth, and gibberellins influence starch accumulation. The interplay of these hormones strongly affects bulking patterns [43].
Importantly, early storage root bulking is highly genotype-dependent. Rüscher et al. (2021) [41] demonstrated that cultivar-specific differences in cambial activation, sugar transporter expression, and phloem/xylem ratios largely dictate how early storage roots develop. Environmental factors, especially soil moisture, temperature, and nutrient availability, may enhance or suppress bulking, but the genetic blueprint remains the primary determinant.
Key Breeding ChallengesCassava breeders face several interconnected constraints, many of which stem directly from the crop’s biology and the complexity of target traits (Table 1). These bottlenecks slow genetic gain and limit the pace of varietal turnover.
 Table 1. Key Breeding Challenges and their implication for breeding.
Table 1. Key Breeding Challenges and their implication for breeding.
The foundation of any successful crop-improvement program lies in the depth and breadth of available genetic resources. Cassava benefits from one of the most geographically and genetically diverse germplasm pools among tropical root crops, owing to its long history of domestication in the Amazon Basin and subsequent diffusion across Africa and Asia. The crop was introduced to Africa in the 16th century and subsequently adapted to a wide range of agro-ecologies, producing thousands of distinct landraces with unique quality, resistance, and culinary properties [46,47].
Today, the conservation, characterization, and utilization of cassava genetic diversity are anchored within several global genebanks and national breeding programs. These collections safeguard not only traditional landraces but also elite breeding lines, wild relatives, and improved varieties that serve as essential materials for breeding, genomic analyses, and prebreeding research.
Major Cassava Global CollectionsInternational Center for Tropical Agriculture (CIAT), Colombia has ~6,000 accessions as the largest international cassava collection, representing broad genetic diversity from South America, the center of origin and diversification. CIAT’s genebank includes wild Manihot species, indigenous landraces, and elite breeding materials, making it a core resource for global prebreeding and genomics research [48].
International Institute of Tropical Agriculture (IITA), Nigeria maintains the most extensive African cassava collection, comprising landraces, farmer-preferred clones, and advanced breeding lines with resistance to cassava mosaic disease (CMD) and cassava brown streak disease (CBSD). IITA materials have been foundational to African varietal improvement and genomic selection initiatives [30].
National Root Crops Research Institute (NRCRI), Nigeria as one of the major custodians of Nigerian cassava diversity, NRCRI holds landraces associated with culturally specific products such as fufu, gari, abacha, and lafun. These accessions are invaluable for improving culinary quality, textural properties, fermentation behavior, and processing efficiency, traits that are increasingly important for consumer- and industry-oriented breeding.
Brazilian Agricultural Research Corporation (EMBRAPA), Brazil houses a vast collection of South American germplasm, including wild relatives, bitter and sweet cassava types, and industrial varieties with diverse starch profiles. These accessions represent the deepest reservoir of allelic diversity for traits such as root architecture, drought tolerance, and starch biochemistry [49].
Together, these collections ensure that cassava breeding programs have access to wide genetic variation essential for addressing emerging challenges, including climate stress, evolving pathogens, changing consumer preferences, and industrial processing demands.
Molecular Diversity StudiesAdvances in molecular markers, particularly SNPs, have transformed the characterization of cassava genetic resources. High-density SNP datasets generated through GBS and SNP arrays have provided new insights into population structure, genetic relationships, and trait-associated alleles across global collections.
Key findings from molecular diversity studies reveal that:
Strong population differentiation exists between African and Latin American germplasm. Because cassava originated in South America, African populations represent a subset of global diversity but have undergone extensive local adaptation. SNP-based analyses consistently show clear genetic partitioning between these regions [23,50].
Deep allelic diversity is found for CMD resistance, carotenoid content, and starch physicochemical properties with several major-effect loci for disease resistance, such as the CMD2 locus, have been identified mainly in African germplasm, while Latin American accessions often harbor unique alleles influencing carotenoids, root dry matter, branching patterns, and cyanogenic potential [17].
Low linkage disequilibrium (LD) across the genome enhances suitability for GWAS; LD in cassava decays rapidly due to outcrossing and high heterozygosity, allowing fine mapping of traits and high resolution in GWAS analyses [50,51]. This property makes cassava one of the more amenable clonally propagated crops for high-resolution genomic dissection.
These molecular insights have accelerated the discovery of markers for quality attributes, yield components, and stress tolerance, ultimately strengthening genomic-assisted breeding pipelines worldwide.
Prebreeding OpportunitiesPrebreeding, introgression of novel alleles from unadapted or wild germplasm into elite breeding pools, is increasingly recognized as a strategic pathway for expanding cassava’s genetic base. Wild Manihot species offer reservoirs of resistance genes, stress-tolerance mechanisms, and value-added biochemical traits not present in cultivated accessions.
Wild Relatives as Sources of Novel DiversityManihot flabellifolia which is widely regarded as one of the closest wild progenitors of cultivated cassava provides alleles for drought tolerance, vigor, and root biomass accumulation. Genetic studies have shown shared ancestry and potential for hybridization with cassava, making it a high-priority species for introgression [47].
Manihot pruinosa and Manihot esculenta ssp. Peruviana are species that harbor genes related to pest and disease resistance, including tolerance to bacterial blight and root rot. They also possess unique starch traits and root morphologies that may be useful for industrial applications such as high-viscosity starch, bioethanol, and biodegradable polymers [52].
Opportunities for Trait EnhancementPrebreeding with wild relatives offers opportunities to introduce: resistance to emerging diseases; improved drought and heat tolerance; novel starch composition and granule morphology; altered cyanogenic glucoside content; enhanced root structural properties for early bulking and mechanization; unique secondary metabolites for biofortification or industrial uses [7,11,14,39].
However, introgression remains challenging due to reproductive barriers, linkage drag, and the need for multiple cycles of backcrossing and genomic selection. Modern tools such as marker-assisted backcrossing, genomic selection, and genome editing may shorten prebreeding cycles and help capture beneficial alleles while minimizing undesirable traits.
Molecular marker technologies have played a central role in transforming cassava breeding from a largely phenotype-driven enterprise into a modern, genomics-assisted discipline. These tools have enabled deeper understanding of the genetic architecture underlying key agronomic, quality, and adaptive traits, while providing the foundation for MAS, GWAS, and GS. The evolution from low-throughput, anonymous markers to high-density, sequence-based platforms have significantly accelerated genetic gain in cassava improvement programs worldwide.
Evolution of Marker SystemsThe application of molecular markers in cassava has evolved over three decades, mirroring advancements in global plant genomics. Each generation of markers addressed specific limitations of its predecessor, ultimately culminating in genome-wide SNP platforms that now underpin modern cassava breeding pipelines.
Early Marker Systems: RAPD and AFLPRandom Amplified Polymorphic DNA (RAPD) and Amplified Fragment Length Polymorphism (AFLP) markers were among the earliest tools used to examine cassava genetic diversity in the 1990s and early 2000s. These markers were relatively inexpensive and required limited prior sequence information, enabling rapid assessment of genetic relationships among landraces and wild Manihot species [53]. However, both systems suffered from limited reproducibility, dominance effects, and low marker density, factors that limited their utility for mapping complex traits.
Simple Sequence Repeats (SSRs): Towards Structured GenotypingThe advent of SSR markers marked a major improvement in cassava molecular genetics. SSRs are co-dominant, highly polymorphic, and more reproducible than RAPD/AFLP markers. They enabled the construction of the first cassava linkage maps, provided the initial insights into population structure, and supported early QTL analyses for yield and disease resistance [54]. SSRs also facilitated germplasm fingerprinting and the management of global genebanks at CIAT, IITA, and EMBRAPA [54].
Single Nucleotide Polymorphisms (SNPs): The Genomic RevolutionThe sequencing of the cassava reference genome [32] marked a turning point in marker development. In the past decade, SNP markers have become the backbone of cassava genomic research and breeding because they offer: high marker density which enables fine-scale dissection of genetic variation; genome-wide coverage suitable for association mapping and genomic prediction; high reproducibility across laboratories and platforms; and scalability for routine use in breeding pipelines, including early-generation selection.
Several SNP platforms now widely support cassava breeding, such as:
Genetic Enhancement of Cassava (GEM) SNP Chip, developed under the NextGen Cassava project, offers thousands of carefully curated SNPs associated with key traits [11].
Diversity Arrays Technology sequencing (DArTseq) provides cost-effective, high-throughput SNP discovery for GWAS, genomic prediction, and diversity studies [51].
Genotyping-by-Sequencing (GBS) is a reduced-representation sequencing approach enabling generation of tens of thousands of genome-wide SNPs [55].
Whole Genome Sequencing (WGS)-derived SNPs is used for high-resolution mapping, evolutionary studies, and identification of structural variants [56–58].
Today, SNP-based genotyping is fully integrated into the breeding pipelines of CIAT, IITA, NRCRI, and EMBRAPA [11,14,16,19]. It forms the foundation of genomic selection, early-generation prediction, and rapid-cycle recurrent selection, strategies critical for increasing the rate of genetic gain in cassava.
QTL Mapping Studies
Quantitative trait loci mapping has been instrumental in identifying genomic regions associated with disease resistance, quality traits, and yield components in cassava. Unlike major-gene crops, cassava improvement requires disentangling complex traits influenced by multiple loci and strong genotype × environment interactions. Marker-based QTL studies provide the first step toward trait dissection, marker-assisted selection, and the design of genomic prediction models.
Disease ResistanceCassava diseases, particularly cassava mosaic disease and cassava brown streak disease, represent major constraints to yield stability and production across Africa. Molecular markers have facilitated significant breakthroughs in understanding and managing these diseases.
Cassava Mosaic DiseaseThe CMD2 and CMD3 loci represent two of the most significant QTL discoveries in cassava disease resistance research.
CMD2 is a dominant resistance locus originally identified in West African landraces. SNP markers tightly linked to CMD2 have allowed reliable marker-assisted selection in breeding programs [30].
CMD3 confers quantitative resistance and complements CMD2 in breeding strategies targeting durable resistance.
The availability of molecular markers for these loci has greatly accelerated the release of CMD-resistant varieties throughout Africa.
Cassava Brown Streak DiseaseQTL mapping and GWAS analyses have identified multiple genomic regions associated with CBSD tolerance, though resistance is more polygenic and environmentally sensitive than CMD. Markers flanking major-effect QTLs on chromosomes IV, VI, and XI have been reported in both CIAT and IITA populations [59]. Integrating these markers into genomic selection increases prediction accuracy for CBSD resistance under high-pressure environments.
Quality TraitsCassava quality traits, including biofortification targets, processing qualities, and nutritional attributes, are increasingly central to breeding objectives. Molecular markers have shed light on the genetic architecture of these following traits in cassava:
Carotenoids (Pro-vitamin A) QTLs associated with β-carotene accumulation have been identified across multiple studies [60]. Markers linked to carotenoid biosynthesis genes such as PSY2 and β-carotene hydroxylase support marker-assisted selection for yellow-fleshed biofortified varieties [61–63].
Several QTLs affecting dry matter, amylose:amylopectin ratio, granule size, and starch viscosity have been mapped, although most show moderate effect sizes. These traits are crucial for end-products such as gari, fufu, and industrial starch derivatives [64–66].
QTLs linked to cyanogenic glucosides on chromosomes II and XVI influence cyanide potential. Markers associated with linamarin and lotaustralin biosynthetic pathways help breeders balance safety, taste, and plant defense [67].
Innovation in Root Trait Phenotyping and Early Bulking ImprovementEarly storage root bulking is a critical trait for market demand, drought escape, shortened production cycles, and farmer adoption across SSA [68,69]. However, the trait is quantitatively inherited, strongly influenced by the environment, and difficult to measure through destructive sampling. Recent advances in functional genomics and high-throughput phenotyping provide opportunities to dissect the genetic basis of early bulking and to model root system architecture in situ. Ground penetrating radar (GPR) provides moderate to high accuracy for non-destructive assessment of cassava below-ground traits. Initial studies showed strong correlations between GPR signals and measured root biomass, with correlation coefficients around r ≈ 0.79 and R² values up to ~0.63–0.77 depending on genotype [35]. Subsequent analyses using improved signal processing and predictive models achieved cross-validated prediction accuracies of r ≈ 0.64–0.72 for root biomass and early bulking indicators [35,70]. These results demonstrate that GPR can reliably capture root volume, architecture, and early bulking dynamics, supporting its use as a high-throughput phenotyping tool in cassava breeding programs.
Yield-related traits remain among the hardest to dissect genetically in cassava due to their strong polygenic control and susceptibility to environmental variation. Studies such as Abah et al. (2024) [71] highlight that QTLs for early bulking, bulking rate, and root size display strong genotype × environment interactions. QTLs identified in one environment often have weak or inconsistent effects elsewhere. Traits such as harvest index, stem number, canopy development, and root number are governed by many small-effect loci distributed across the genome [11,16].
Because bulking and yield traits are highly polygenic, they are ideal targets for genomic selection, which captures genome-wide additive variation rather than relying on individual marker effects.
Advances in cassava genomics over the past decade have radically improved our understanding of the crop’s biology and opened new frontiers for genetically informed breeding. The availability of a chromosome-level reference genome, coupled with functional genomic tools such as transcriptomics, epigenomics, and regulatory network analyses, allows researchers to dissect complex traits, including early storage root formation, disease resistance, and starch quality, at molecular resolution. These resources have significantly strengthened the integration of genomics into cassava breeding programs.
The Cassava GenomeThe release and continuous improvement of the cassava reference genome have been transformative for cassava biology. The current assembly (v8.1) provides one of the most complete and accurate representations of the cassava genome to date, offering ~750 Mb genome assembly, capturing the majority of euchromatic and heterochromatic regions; approximately 33,000 predicted protein-coding genes, reflecting cassava’s high allelic diversity and genomic complexity; chromosome-level scaffolding, enabling precise gene localization, structural variant identification, and comparative mapping across Manihot species [14,32]. This high-quality assembly supports diverse downstream applications, including GWAS, QTL mapping, genomic selection, and evolutionary studies.
Key Gene Networks Uncovered Through Genomic AnalysesThe reference genome has significantly improved gene discovery for multiple biochemical and physiological pathways central to cassava improvement:
In starch biosynthesis pathway, genes underpinning starch production and functionality, including granule-bound starch synthase I (GBSSI), starch branching enzymes (SBE1/2), and soluble starch synthase II (SSII), have been fully annotated. These genes explain variation in amylose content, pasting viscosity, gelatinization temperature, and suitability for industrial applications [72,73].
In cyanogenic glucoside pathway, the biosynthesis of linamarin and lotaustralin is governed by cytochrome P450 genes CYP79D1, CYP79D2, and downstream enzymes such as hydroxynitrile lyase (HNL). These genes have become major targets for engineering low-cyanide cassava and improving food safety [74].
Study on carotenoid accumulation, showed that biofortification efforts have benefited greatly from genomic identification of genes regulating β-carotene levels, including phytoene synthase (PSY), orange (OR), and β-carotene hydroxylase. These loci underpin the development of yellow-fleshed cassava with enhanced vitamin A content [75,76].
Under the root initiation and bulking regulatory networks, genomic analyses have revealed transcription factors, hormone signaling components (auxin, cytokinin, ABA), and cell-wall modifying enzymes that influence root initiation, secondary growth, and early bulking. Key regulators include MCM1, AGAMOUS, DEFICIENS, SRF (MADS)-box genes, NAM (No Apical Meristem), ATAF1/2, CUC2 (NAC)-domain proteins, expansins, and sugar transporters, elements crucial for early storage root formation. Together, these genomic discoveries provide unprecedented opportunities to manipulate trait pathways through MAS, genomic selection, and targeted gene editing [11,16,19].
TranscriptomicsTranscriptomics, especially RNA sequencing (RNA-seq), has become one of the most powerful tools in cassava functional genomics. By profiling gene expression under different developmental stages, environments, and stress conditions, researchers can elucidate the molecular mechanisms regulating complex traits.
Insights from Cassava Transcriptome StudiesOn Early Storage Root Initiation Pathways; RNA-seq analyses comparing fibrous roots, initiating storage roots, and bulking roots reveal shifts in expression of genes associated with: cambial activation; starch biosynthesis; sugar transport [Sugars Will Eventually be Exported Transporters (SWEET) and Sucrose Transporters (SUT)]; phytohormone biosynthesis and signaling. These expression signatures highlight candidate genes that may control the timing and intensity of early bulking, a critical breeding target [41].
Drought Response Regulatory Modules: Cassava exhibits remarkable resilience to episodic drought. Transcriptomic profiling under water stress conditions has identified: ABA-responsive transcription factors; late-embryogenesis abundant (LEA) proteins; ROS-scavenging enzymes; aquaporins and osmotic adjustment proteins. These modules reveal how cassava maintains carbon assimilation and root growth under fluctuating moisture regimes [77,78].
CMD Resistance Transcriptional Networks: Comparative transcriptomics between CMD-resistant and susceptible genotypes show differential expression of: NBS-LRR resistance genes; RNA silencing components; viral replication inhibitors; cell wall–associated defense genes. Insights from these studies complement genetic mapping of CMD2/CMD3 and help clarify the molecular basis of durable resistance [79–81].
Postharvest Physiological Deterioration (PPD) and Cooking Quality: PPD, a major challenge affecting fresh root shelf life, is associated with rapid oxidative bursts and activation of stress-related genes. Transcriptomic studies have identified: peroxidases; phenolic pathway enzymes; programmed cell death regulators. Cooking quality studies show differential expression of genes controlling cell wall softening, starch gelatinization, lignification, and water binding capacity, all critical in traits such as fufu texture, gari expansion, and boiling time [82,83].
Transcriptomics as a Breeding AcceleratorThe integration of transcriptomic data with genomic markers, metabolomics, and phenomics enables: identification of predictive expression markers; mapping of regulatory networks; development of gene-based selection indices; improved genomic prediction accuracy; discovery of candidate genes for Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-based editing.
Thus, transcriptomics now plays a central role in modern cassava improvement strategies, bridging the gap between genotype and phenotype [84].
Genome-wide association studies have become one of the most useful tools in cassava genetics, enabling the dissection of complex, quantitatively inherited traits at high resolution. Cassava’s reproductive biology, namely, its obligate outcrossing system and high heterozygosity, results in rapid linkage disequilibrium decay across the genome. This unique genomic characteristic allows GWAS to pinpoint loci with greater precision than in many other clonally propagated crops [50]. The widespread adoption of high-density SNP genotyping platforms such as GBS, DArTseq, and GEM SNP chips has further expanded the power of GWAS across diverse breeding populations.
GWAS has now been applied to a wide range of agronomic, nutritional, physiological, and disease-resistance traits, uncovering genetic variants of direct relevance to breeding programs at CIAT, IITA, NRCRI, EMBRAPA, and other global partners [17,23,71].
Why GWAS Works Well in CassavaSeveral genomic and biological features make cassava particularly suited for GWAS:
As a result of low to moderate LD or rapid LD decay; LD in cassava declines rapidly over short genomic distances due to frequent outcrossing. This allows researchers to identify loci with fine-scale resolution, narrowing candidate gene lists dramatically [49].
As a result of high allelic diversity; extensive diversity in landraces and breeding populations increases the probability of capturing functional alleles associated with traits of interest [85,86].
Availability of large, structured breeding populations; Multi-family, multi-environment datasets developed by CIAT, IITA, and NextGen Cassava provide robust phenotypic and genotypic resources for association mapping [87].
As a result of chromosome-level reference genome; The V8.1 assembly allows precise anchoring of significant SNPs and facilitates candidate gene discovery.
Combined, these features support high-confidence mapping of traits governed by many small-effect loci, traits traditionally considered “intractable” under classical breeding.
Key GWAS Discoveries in Cassava β-Carotene Accumulation and Nutritional BiofortificationGWAS studies have successfully identified major loci associated with carotenoid accumulation, crucial for developing biofortified cassava to combat vitamin A deficiency. Ikeogu et al. (2009) [88] identified SNPs linked to PSY2, ORANGE (OR), and downstream carotenoid pathway genes. These discoveries enabled genomic selection models that predict carotenoid content with high accuracy and supported marker-assisted selection for yellow-fleshed varieties. These markers have already been integrated into biofortification pipelines at IITA and HarvestPlus.
Disease Resistance (CMD and CBSD)Genome-wide association studies have significantly advanced the molecular understanding of cassava’s two most devastating viral diseases: Cassava Mosaic Disease and Cassava Brown Streak Disease. For CMD, GWAS has consistently identified strong associations on Chromosome 12, corresponding to the well-characterized CMD2 locus, as well as additional quantitative resistance regions on Chromosome 16 [17]. These loci have been shown to contribute to durable, broad-spectrum resistance, complementing earlier QTL mapping studies and providing molecular targets for marker-assisted selection. Analyses of these regions reveal enrichment of functional transcripts related to key defense mechanisms, including Nucleotide-Binding Site-Leucine-Rich Repeat (NBS-LRR) resistance genes, components of the RNA-silencing machinery, and other antiviral effectors. Such genes form part of the cassava innate immune system, enabling recognition of viral pathogens and activation of downstream defense responses. In contrast, CBSD resistance exhibits a more polygenic architecture and is highly influenced by environmental conditions, making it more complex to dissect genetically. Nevertheless, GWAS analyses have successfully identified consistent SNP markers associated with CBSD resistance, often located near genes implicated in cell-wall signaling, antiviral responses, and root-specific defense mechanisms [89]. These findings highlight the role of multiple, small-effect loci in conferring partial resistance and underscore the importance of integrating genomic information across diverse environments for breeding CBSD-resistant varieties.
Starch Quality, Dry Matter Content, and Physicochemical TraitsGWAS has identified SNPs associated with starch composition, including amylose:amylopectin ratios, starch branching enzymes, granule size, dry matter content, and hydration/pasting behavior [16,90]. These loci are directly relevant to industrial processing (starch, ethanol), food-texture traits (fufu moldability, gari expansion), and improved shelf stability. The findings enable breeders to select for complex quality traits, integrating functional genomics with phenotypic data to enhance both product quality and processing efficiency.
Early Storage Root BulkingEmerging GWAS analyses have begun revealing early bulking markers associated with: sugar transport (SWEET gene family); cambial activity regulators; hormone signaling pathways; carbohydrate partitioning networks.
Although effect sizes are typically small and environmentally variable, the identification of these markers provides useful biological insight and supports development of genomic prediction models tailored for early bulking [71].
Multi-Environment GWAS: A Major AdvancementCassava traits often display strong genotype × environment (G × E) interactions, especially disease scores, root quality, and bulking traits. Single-environment GWAS analyses frequently fail to detect stable, robust loci.
Benefits of Multi-Environment GWAS ApproachesCombining multi-environment datasets has brought several key benefits for cassava breeding:
It helps to increased power and reliability; by combining phenotypic and genotypic data across diverse locations, researchers can detect loci that remain stable under variable disease pressure, climate, and soil conditions. This approach reduces environmental noise and improves the confidence in identified marker–trait associations. Stable loci across environments provide reliable targets for selection [91].
It helps in the identification of environment-specific alleles; multi-environment trials reveal alleles that are expressed or show significant effects only under certain stresses or agro-ecological conditions, such as drought, nutrient limitation, or high disease prevalence. These environment-specific alleles capture adaptive responses and inform breeding for stress resilience [17].
It provides improved integration with genomic prediction; loci that demonstrate stability across environments contribute consistent signals to genomic prediction models, increasing the accuracy of genomic estimated breeding values (GEBVs) for traits of interest. Multi-environment GWAS and mixed-model approaches that account for genotype × environment interactions enhance predictive performance compared with single-environment analyses. [22,92].
GWAS as a Breeding AcceleratorThe integration of GWAS into breeding pipelines has produced several practical outcomes: enables discovery of candidate genes for functional validation; supports MAS for major-effect loci (CMD2, carotenoid genes); feeds trait-associated SNPs into genomic selection models; refines breeding population design and parental selection; accelerates genetic gain for polygenic traits [17,22,70].
With increasing availability of multi-omic data, transcriptomics, metabolomics, and phenomics, future GWAS approaches may adopt multi-omic GWAS and causal modeling to uncover deeper regulatory layers.
Genomic selection (GS) has emerged as one of the most transformative innovations in cassava improvement, enabling breeders to predict the genetic potential of individuals using genome-wide markers before phenotypic evaluation is completed. This approach is particularly impactful for cassava, where long growth cycles, clonal propagation, and strong genotype × environment (G × E) interactions slow down gains from traditional breeding. By capturing effects from thousands of small-effect markers simultaneously, GS accelerates the identification of superior clones and supports rapid-cycle breeding pipelines.
GS was first implemented in cassava through the collaborative efforts of the NextGen Cassava Breeding Project, integrating genomic datasets from CIAT, IITA, NRCRI, and national programs across Africa and Latin America. Today, GS is fully embedded in elite breeding pipelines and has demonstrably increased genetic gains for yield, resilience, and quality traits [17].
Applications of GS in Cassava Root Yield and Productivity TraitsYield is one of the most complex traits in cassava, controlled by numerous loci of small effect and heavily influenced by environmental variation. Traditional QTL-based approaches explain only a small portion of yield variance. GS overcomes this limitation by using all genome-wide SNPs to predict breeding values.
Wolfe et al. (2017) [11] demonstrated that GS achieves moderate-to-high prediction accuracy for root yield, harvest index, and related productivity traits.
GS enables selection among seedlings or early-stage clones without waiting for full clonal evaluations, dramatically speeding up genetic gains.
Yield-related GS models have been particularly effective when trained on multi-year, multi-location datasets, allowing better modeling of G × E patterns.
Cassava Mosaic Disease (CMD) ResistanceAlthough the major loci CMD2 and CMD3 confer strong resistance to cassava mosaic disease (CMD), the observed phenotypic expression of CMD resistance still exhibits quantitative variation among progenies. This variation arises from the contributions of multiple minor-effect loci, background genetic variation, and genotype × environment interactions. Genomic selection (GS) has become a key tool in breeding programs to manage this complexity. By using genome-wide marker data, GS enables the prediction of CMD resistance in progenies even before they experience significant disease pressure, allowing early and accurate selection of promising clones. Additionally, GS facilitates the combination of major-effect CMD loci with minor background resistance alleles, supporting the development of durable and broad-spectrum resistance that is less vulnerable to pathogen evolution. Another advantage of GS is that it helps resolve confounding effects between CMD resistance and growth-related traits during clonal selection, allowing breeders to select for resistance without inadvertently selecting against yield or other agronomic traits. This integration of genome-wide information with breeding strategies has been demonstrated to improve selection efficiency and accelerate genetic gain for CMD resistance in cassava breeding programs [93].
Root Quality Traits (Dry Matter, Starch, Carotenoids)Quality traits are essential for consumer preference, processing properties, and nutrition, show high heritability and strong genetic signals. GS has proven effective for: dry matter content (DM) as a key trait for gari, fufu, and industrial starch; starch viscosity and physicochemical properties; carotenoid content, enabling rapid advancement of biofortified yellow cassava. Combining genomic and metabolomic data further improves prediction accuracy for complex starch and quality traits [94].
Multi-Environment Genomic PredictionsCassava is cultivated across diverse agro-ecologies with varying disease pressure, rainfall patterns, soil fertility, and climate regimes. To address these challenges: Multi-environment GS models incorporate G × E interactions directly into predictions; Reaction-norm models, genomic genotype-by-environment models, and Bayesian hierarchical approaches significantly improve prediction accuracy; These models have been particularly valuable for yield, CBSD responses, and drought-related traits. This approach strengthens varietal stability and supports more targeted deployment recommendations [95].
Benefits of Genomic SelectionGS offers several strategic advantages that align exceptionally well with cassava’s biology and breeding system.
Shortened Breeding Cycles (Reduction by 2–4 Years)Cassava’s long growth cycle traditionally limits breeding progress. GS: allows early selection at seedling or nursery stages; reduces the number of clonal evaluation cycles; enables rapid-cycle recurrent selection. Breeding programs that once completed a cycle in 8–10 years can now complete cycles in 4–6 years.
Increased Selection IntensityBecause GS allows selection from large seedling populations without the need for extensive early phenotyping: breeders can screen thousands of genotypes cheaply using DNA alone; only the genetically superior clones advance to field testing; selection intensity increases substantially, improving genetic gain per cycle. This is especially important for elite × elite crosses targeting yield, CMD resistance, and root quality.
Improved Accuracy for Polygenic TraitsTraits such as yield, bulking rate, dry matter content, and stress resilience are governed by many loci with small individual effects. GS excels in these contexts because: it captures genome-wide additive and non-additive variance; it avoids the limitations of QTL-based MAS; it improves prediction accuracy even when individual markers explain small portions of variance. Thus, GS is a natural fit for cassava’s most challenging traits.
Seamless Integration with Clonal PropagationCassava’s vegetative propagation system supports GS in several ways: once superior clones are identified via genomic prediction, they can be propagated and tested quickly; genomic predictions made early in the pipeline directly translate into clonal performance; multi-stage clonal trials can be reorganized to focus resources on genetically superior candidates. This integration enhances both efficiency and resource use.
Supports Modern Breeding ObjectivesGS also complements other cutting-edge innovations like high-throughput phenotyping (HTP) for root architecture and canopy traits; multi-omic prediction models that combine SNPs, transcripts, and metabolites; gene editing by identifying high-value genetic targets; accelerated parental selection enabling more frequent crossing cycles. Through these synergies, GS strengthens the entire cassava breeding ecosystem.
Recent advances in high-throughput “omics” technologies have opened new possibilities for understanding cassava biology at molecular, biochemical, and systems levels. Because cassava traits such as storage root bulking, disease resistance, postharvest quality, and nutritional attributes are multi-layered and polygenic, single-omic approaches often provide only partial insight. Multi-omics integration, combining metabolomics, transcriptomics, proteomics, genomics, and phenomics, offers a holistic framework to uncover regulatory mechanisms and accelerate precision breeding.
These tools enable breeders and biologists to link genotype to phenotype more accurately, identify biomarkers, model regulatory networks, and predict complex trait performance with greater confidence. Multi-omics approaches are now central to modern cassava improvement programs, particularly through initiatives such as NextGen Cassava, HarvestPlus, CGIAR Excellence in Breeding, and national programs at NRCRI, IITA, and CIAT [96].
MetabolomicsMetabolomics has emerged as a strategic molecular phenotyping platform in cassava research because metabolites represent the end-products of biological regulation and therefore define the direct biochemical phenotype of a genotype. Unlike DNA markers or transcripts, which indicate potential or regulatory activity, metabolites reflect the realized physiological and biochemical state, providing the most proximal measurable layer linking gene function to expressed agronomic, nutritional, and processing traits [97]. Cassava storage roots are biochemically rich sink organs characterized by large carbon flux, starch accumulation, cyanogenic compounds, soluble sugars, and wide metabolic plasticity, making metabolomics particularly well-suited for interrogating complex quality traits, toxicity reduction, stress physiology, and end-use properties relevant to consumer and industrial priorities [14,98].
Transcriptome-to-metabolome associations have shown that metabolic traits can vary even among genotypes that share similar QTL profiles, due to rapid post-transcriptional regulation and environmental responsiveness [99]. This reinforces the importance of metabolomics for breeding decisions in cassava improvement pipelines, most especially for traits where biochemical composition defines product identity more strongly than agronomic appearance alone [100]. Cassava breeding programs now increasingly rely on metabolomic readouts to capture traits such as starch behavior, sugar partitioning, cyanide potential, volatile compound diversity, root aging physiology, and stress-induced metabolic resilience, all of which are critical to end-user acceptability and variety adoption [101].
One of the most impactful areas of metabolomics application in cassava is the characterization of storage-root starch structure and its physicochemical behavior under processing. By profiling biosynthetic intermediates associated with amylose and amylopectin assembly, metabolomics enables differentiation of starch granule branching density, chain-length distribution, crystalline lamella arrangement, peak viscosity, pasting temperature, and gelatinization dynamics. These parameters directly influence sensory texture and final product behavior in major cassava-based foods, including fufu, gari and industrial starch forms [101,102]. Metabolomic profiles showing elevated root-localized carbon intermediates of the ADP-Glucose pathway have consistently correlated with increased storage-root bulking efficiency and improved starch cohesive behavior during dough formation, key parameters that determine fufu moldability, elasticity, and product texture perception [103]. These biochemical measurements support breeders in selecting genotypes that produce starch with optimal swelling, dispersibility, retrogradation patterns, and paste stability required for both fermented cassava foods and starch-industry utility [103].
Metabolomics equally supports quantitative resolution of sugar profiles, offering insight into carbon partitioning efficiency and sink strength in storage-root initiation. Comparative carbohydrate pool analyses (glucose, fructose, sucrose, and maltose levels) have allowed characterization of carbon allocation dynamics between source tissues (leaves) and sink organs (roots). Genotypes with high sucrose abundance in storage-root initials exhibit higher sink strength, increased phloem unloading efficiency, and accelerated root conversion from fibrous to bulking state, thereby promoting earlier bulking initiation and increased fresh-root biomass yield [104]. Stable sugar assays using Gas Chromatography–Mass Spectrometry (GC-MS) have been widely validated as biochemical predictors for selecting cassava lines that demonstrate optimal carbon flow into roots and yield-stability under fluctuating climatic or agronomic environments, offering critical value in juvenile-phase selection during breeding cycles [100].
Efforts to reduce cassava toxicity have also been advanced by metabolomics through direct measurement of cyanogenic glucosides, primarily linamarin and lotaustralin. These aliphatic nitrile-linked secondary metabolites represent the major cyanide sources in cassava, and their quantification by metabolomic platforms allows for high-precision estimation of total cyanide potential (TCP) across genotypes, environments, and processing states. This precision is essential when breeding for varieties suitable for direct human consumption or fast processing, where cyanide thresholds determine safety compliance [98,101]. In West African breeding scenarios, metabolomic cyanide mapping has significantly improved the ability of breeding programs to prioritize low-linamarin genotypes without sacrificing root size or dry-matter content, a balance that is otherwise difficult to achieve using agronomic selection alone [14,105]. These safety-guided metabolite screens are now viewed as essential complementary analyses alongside field-based dry-matter or fresh-root yield data.
Metabolomics has also progressed the molecular interpretation of consumer-driven quality traits. Storage-root metabolite profiling has supported characterization of volatile organic compounds (VOCs), short- and long-chain fatty acids, organic acids (lactic, malic, citric, succinic), aldehydes, esters, ketones, alcohols, phenolics and flavor-conferring secondary metabolites that collectively define taste, aroma, after-fermentation acidity, dough collapse resistance and textural perception. These biochemical traits influence fufu aroma intensity, elasticity, dough stretchability, moldability, stickiness, taste complexity and user-perceived cooking quality [101]. Sensory metabolomics studies showed that cassava genotype-specific VOC clusters can distinguish consumer-preferred organoleptic profiles for fermented cassava food products, supporting biochemical trait selection for variety-adoption breeding cycles [100]. Moreover, metabolomics-sensory correlation models allow screening for secondary metabolites (fermentation-related volatiles, polyols, phenols) that both enhance eating quality and minimize off-aromas or textural collapse during dough shaping, helping breeding programs better predict gastronomic acceptability through biochemical fingerprinting rather than field appearance alone [99].
Because metabolites respond dynamically to both genotype and the environment, metabolomics equally supports modeling of genotype × environment × metabolite (G × E × M) interactions. These interactions enhance understanding of environment-sensitive processes including early bulking response to rainfall or dry seasons, metabolite fluctuation under drought or virus pressure, browning sensitivity after harvest, and metabolite-induced variability during fermentation-or-processing phases. The ability to characterize these G × E × M interactions allows breeding programs to identify metabolic stability markers for robust varietal screening under climate uncertainty [41,104]. Thus, beyond quality trait characterization, metabolomics supports cassava breeding by monitoring biochemical consistency, root aging physiology, food-metabolite compliance, stress memory pathways, and metabolic vigor that translate into reliable enterprise performance for women-led processing units dependent on cassava product quality in real-world settings [106,107].
Metabolomics provides cassava breeding with the ability to fingerprint biochemical identity of storage-root starch or sugar architecture, measure cyanide potential to ensure product compliance, characterize VOC and flavor-conferring molecule clusters that determine fufu or gari quality, and model G × E × M systems for climate-responsive variety stability. These contributions push cassava breeding beyond indirect markers toward biochemical phenotype-first varietal improvement models that enhance both consumer-preferred qualities and industrial starch adoption [99].
TranscriptomicsTranscriptomics, largely enabled by RNA sequencing technology (RNA-seq), offers genome-wide resolution of transcriptional activity, allowing researchers to quantify and compare gene expression dynamics across tissues, developmental stages, environmental conditions, and contrasting genotypes. In cassava, transcriptomic resources have played a foundational role in decoding regulatory circuits underlying storage-root formation, disease resistance, abiotic stress responses, and nutritional improvement. These datasets bridge molecular biology and breeding by revealing not only differentially expressed genes but also gene co-expression modules, pathway activation hierarchies, and regulatory hubs that coordinate complex trait expression [99].
One of the earliest applications of transcriptomics in cassava bulking research focused on distinguishing fibrous (non-storage) roots from storage-root initials. RNA-seq comparisons demonstrated that early bulking is marked by upregulation of gene families involved in cambial and meristematic reactivation, including members of the MADS-box, NAC, and WOX transcription factor families, which promote vascular cell identity and secondary root thickening. These studies further revealed heightened transcription of sucrose transporters (including SUT and SWEET gene clades), starch assembly enzymes, and hormone-responsive regulators integrated in auxin, abscisic acid (ABA), and cytokinin signaling pathways, collectively defining the molecular shift from exploratory to storage sink behavior [41]. The identification of these bulking-associated expression signatures has provided a template for modeling source-to-sink transitions and for creating transcriptional biomarkers that inform early selection in breeding populations [41].
Cassava mosaic disease resistance has also been clarified through differential transcriptome analyses between resistant genotypes carrying CMD2 or CMD3 loci and susceptible lines. These studies showed strong transcriptional enrichment of activation-induced immune receptors, particularly NBS-LRR (nucleotide-binding site leucine-rich repeat) and LRR-RLK (receptor-like kinase) sensors, which activate patterned antiviral signaling. Transcriptomics also uncovered high expression of RNA interference (RNAi) pathway genes, including DCL (Dicer-like), RDR (RNA-dependent RNA polymerase), and AGO (Argonaute) families that regulate antiviral small-RNA biogenesis. In addition, defense-primed genes contributing to pectin methylesterification, callose deposition, and cell-wall fortification were observed to support resistance containment and durability [108,109]. These mechanistic findings have complemented trait loci detected through QTL or GWAS by explaining how resistance alleles shape downstream immunity cascades for more durable viral defense [104].
Under drought and prolonged water-limitation conditions, cassava mounts a layered transcriptional response dominated by ABA-responsive regulons. Transcriptome studies identified dehydration-induced activation of hormonal receptors [Pyrabactin Resistance/ PYR1-Like/ Regulatory Component of ABA Receptor (PYR/PYL/RCAR)], transcriptional regulators [ABA-Responsive Element Binding protein/ ABRE-Binding Factor (AREB/ABF)], osmolyte and osmoprotectant biosynthesis pathways (including raffinose, trehalose, and late-embryogenesis abundant proteins), and antioxidant enzyme gene clusters [Superoxide Dismutase (SOD), Catalase (CAT), Ascorbate Peroxidase (APX), Glutathione Peroxidase (GPX)] that protect cells from induced oxidative burst. WRKY, Myeloblastosis (MYB), Basic Leucine Zipper (bZIP), and Basic Helix-Loop-Helix (bHLH) transcription factor families were consistently reported as central regulators of stomatal modulation, cellular homeostasis, sugar reallocation, and gene network reinforcement under drought stress. These integrated pathways explain the molecular basis of cassava’s unique ability to maintain productivity under low water environments [104,110].
Cassava biofortification efforts, particularly breeding for enhanced β-carotene content, have equally benefited from transcriptomics. Deep expression profiling of carotenoid pathways revealed structural gene upregulation, including phytoene synthase (PSY), orange (OR) chaperones, carotenoid cyclases [Lycopene β-Cyclase/ Lycopene ε-Cyclase (LCYB/LCYE)], and plastid-localized transport regulators that influence carotenoid sequestration and stability. Weighted gene co-expression network analysis (WGCNA) has identified metabolic modules linking PSY-OR co-activation with downstream carotenoid assembly networks, providing clear path for nutritional trait prediction and marker development for biofortified breeding pipelines [111]. The ability to decipher such co-regulated networks enables breeders to shift away from single-gene models toward coordinated pathway‐level improvement, which is especially important for heterozygous crops such as cassava [99].
Collectively, transcriptomics has empowered cassava breeding by enabling: identification of developmental switches in early storage-root bulking; clarification of defense regulons underlying CMD resistance; resolution of hormone-driven drought stress signaling networks; and mapping of pathway-co-expression modules that improve nutritional biofortification. These advances demonstrate that cassava improvement increasingly depends on multi-layered gene network interpretation rather than isolated genes, providing the biological context necessary to design superior genomic selection models for root yield, disease durability, industrial starch profile, and climatic resilience [99,104].
ProteomicsProteomics provides insight into functional proteins, the molecular machines executing biochemical pathways. Because proteins undergo post-translational modifications and are not always predictable from transcript levels, proteomics fills critical gaps left by other omics layers. Proteomics has made significant contributions to understanding cassava biology, especially with respect to root development, starch metabolism, hormonal regulation, and stress responses. For instance, comparative proteomic studies during cassava storage-root development have identified many enzymes involved in carbohydrate metabolism and starch accumulation, including active isoforms of starch-branching enzymes and other starch-synthesis related proteins, underscoring the biochemical control of starch architecture in developing roots [31]. Beyond starch metabolism, proteome analyses of cassava roots at different developmental stages have revealed shifts in proteins linked to energy production, cell structure, and stress defense, illustrating the complexity of metabolic reprogramming during storage-root formation [112].
Moreover, proteomic investigations under abiotic stress conditions, for example drought, have documented alterations in the abundance of stress- and defense-related proteins, including antioxidant enzymes and proteins involved in ROS detoxification. These findings highlight potential molecular mechanisms by which cassava adjusts its physiology under water deficit, offering candidate targets for breeding drought-resilient varieties [110]. Proteomics thus provides a functional, protein-level view of the cellular machinery underlying root growth, starch deposition, and stress responses in cassava, insights that go beyond gene expression studies and help bridge genotype to phenotype for traits of agronomic importance such as yield, starch quality, and stress resilience.
Integrated Platforms: Multi-Omics Fusion for Precision BreedingThe power of multi-omics lies not in individual datasets but in their integration. When genomics, metabolomics, transcriptomics, proteomics, and phenomics are fused within systems biology frameworks, they enable unprecedented clarity into the genotype–phenotype continuum.
Benefits of Multi-Omics IntegrationThe integration of multi-omics datasets offers several significant benefits for cassava breeding and the understanding of complex traits. One major advantage is biomarker discovery, as integrative analyses can identify metabolites, genes, or protein signatures that are predictive of traits such as early bulking, post-harvest physiological deterioration (PPD) resistance, cassava mosaic disease (CMD) immunity, or cooking quality. These biomarkers can accelerate breeding decisions and enhance the accuracy of selection by providing measurable indicators of desired phenotypes.
Multi-omics integration also facilitates the identification of regulatory hubs within biological networks. Network modeling can reveal master regulators, including transcription factors, signaling molecules, and hormonal nodes that control multi-gene pathways. These hubs represent prime candidates for gene editing, offering targeted opportunities to influence complex trait expression.
Moreover, integrating omics datasets can provides mechanistic insights into complex traits by uncovering causal pathways that link gene expression, metabolic flux, and phenotypic outcomes. This understanding is particularly critical for polygenic traits, where the interplay of multiple genes and pathways determines the final phenotype.
Enhanced genotype–phenotype associations are another key benefit. Approaches such as multi-omic genome-wide association studies (GWAS) and multi-kernel genomic prediction models combine multiple layers of biological information, improving the ability to detect trait-associated loci and increasing prediction accuracy.
Multi-omics integration opens new pathways for climate-resilient breeding. Traits such as drought tolerance, heat resilience, and disease resistance can be modeled as interconnected, multi-layered biological systems rather than as isolated features. This holistic view enables the development of more robust cassava varieties that are better adapted to environmental stresses, supporting sustainable crop production in the face of climate change.
The rapid development of genomic tools and biotechnologies promises to transform cassava breeding, enhancing the crop’s potential to contribute to food security, nutrition, climate resilience, and socio-economic development. One of the key drivers of future cassava improvement is the application of genomic selection (GS) and marker-assisted selection (MAS), facilitated by the increasing availability of high-density molecular markers such as SNPs and SSRs, as well as high-throughput genotyping and comprehensive genome data [113,114]. Genomic selection enables breeders to predict breeding values based on genomic information, reducing the need for lengthy and costly field trials, a particularly important advantage given cassava’s long growth cycle and highly heterozygous genome [113]. Large-scale genome resequencing efforts, which have analyzed hundreds of cassava accessions and identified thousands of high-quality SNPs and candidate loci for critical agronomic traits, further enable mapping and selection for complex traits such as yield stability, root quality, stress tolerance, and heterozygosity management [114].
Advances in gene editing and molecular biotechnology also offer unprecedented opportunities for targeted trait improvement in cassava. The advent of precise genome editing tools, most notably CRISPR/Cas9, allows the development of cassava varieties with enhanced disease resistance, improved nutritional profiles, superior starch quality, reduced cyanogenic glucoside content, and delayed post-harvest deterioration, traits that are difficult or slow to achieve through conventional breeding [114]. Gene editing can enable the removal or modification of susceptibility genes or metabolic pathways to reduce cyanide content or improve starch composition [114]. Complementing these techniques, “omics”-based functional genomics, including transcriptomics, proteomics, and metabolomics, provides detailed insights into the molecular basis of stress tolerance, root quality, starch biosynthesis, and other complex traits, thereby supporting more precise and informed breeding decisions [113,114]. These integrated genomic and functional approaches are likely to be pivotal in developing cassava varieties that meet both agronomic requirements and consumer demands.
In addition, new breeding strategies are expected to shorten breeding cycles and increase genetic gain. Inbred-parent–based hybrid breeding strategies have been proposed to overcome challenges related to heterozygosity and long generation times [113]. When combined with genomic selection and advanced genotyping, these strategies could significantly reduce breeding cycles, improve trait introgression, eliminate deleterious alleles, and exploit heterosis. Such approaches promise to accelerate the development of cassava varieties that are resilient to climate variability, resistant to emerging pests and diseases, and better aligned with evolving market demands [114].
The effectiveness of genomics-assisted breeding also depends on robust data management and high-throughput phenotyping. Digital platforms such as CassavaBase allow breeding programs to efficiently manage large datasets, including phenotyping, genotyping, and crossing information, thereby improving quality control and accelerating decision-making [115]. High-throughput phenotyping methods, including near-infrared spectroscopy, drone- or imaging-based field assessment, and automated root quality evaluation, are likely to become increasingly widespread [116]. These technologies help overcome bottlenecks associated with slow, labor-intensive, and error-prone manual phenotyping, particularly for complex traits such as yield stability, drought tolerance, root quality, and post-harvest resilience.
Collectively, these advances suggest a future in which genomics-assisted cassava breeding can deliver high-yielding, nutrient-rich, and climate-resilient varieties more rapidly and efficiently than ever before. By integrating genomic tools, molecular biotechnology, advanced breeding strategies, and digital phenotyping platforms, cassava breeding programs are positioned to transform both the crop and the livelihoods of the millions who depend on it, particularly in regions vulnerable to food insecurity and climate stress [113,114] as shown in Figure 1.
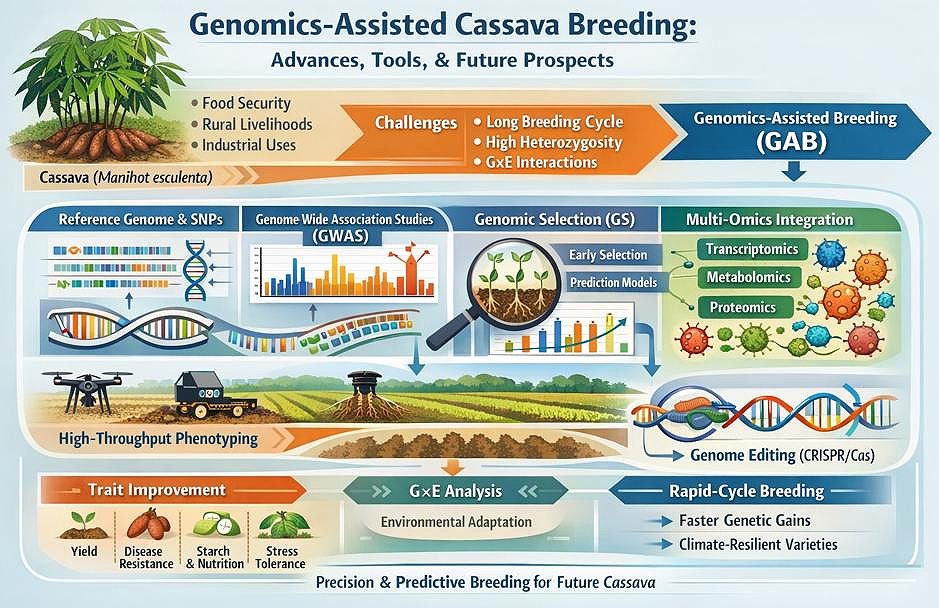 Figure 1. The future of quality cassava production through precision and predictive breeding.
Figure 1. The future of quality cassava production through precision and predictive breeding.
Genomic selection (GS) and multi-omics integration are transforming cassava breeding by providing tools to accelerate genetic gain, improve accuracy in complex trait prediction, and optimize resource allocation. GS enables earlier selection at seedling or nursery stages, shortens breeding cycles by 2–4 years, and increases selection intensity, particularly for polygenic traits such as yield, dry matter content, and stress resilience. Its seamless integration with clonal propagation ensures that genetic predictions translate directly into field performance, allowing breeders to focus resources on the most promising genotypes.
The integration of metabolomics, transcriptomics, proteomics, and other omics layers provides a comprehensive view of cassava biology, linking genotype to phenotype with unprecedented precision. Metabolomics enables the profiling of storage-root biochemical traits, cyanogenic glucoside content, starch architecture, sugar partitioning, and volatile organic compounds that determine consumer-preferred qualities and processing suitability. Transcriptomics reveals regulatory networks controlling storage-root initiation, drought tolerance, CMD resistance, and biofortification traits, while proteomics uncovers functional proteins that execute biochemical pathways and stress responses.
Multi-omics integration enhances the discovery of biomarkers, regulatory hubs, and causal pathways, thereby improving selection accuracy and enabling precision breeding for complex traits. These approaches also support the development of climate-resilient cassava varieties, capable of maintaining yield, quality, and stress tolerance under fluctuating environmental conditions. Advances in gene editing, high-throughput phenotyping, and digital breeding platforms further complement these innovations, offering the potential to accelerate breeding cycles, reduce labor-intensive field evaluation, and optimize trait introgression.
The combination of GS, multi-omics, and modern breeding strategies positions cassava improvement programs to meet both agronomic and industrial demands. These approaches facilitate the development of high-yielding, nutritious, safe, and climate-resilient cassava varieties, thereby contributing to food security, economic development, and the livelihoods of millions who depend on this staple crop. The integration of molecular and systems-level understanding into practical breeding pipelines represents a paradigm shift, moving cassava improvement from conventional phenotype-based selection toward predictive, data-driven, and precision-oriented strategies.
No new data were generated or analyzed in this study. All data discussed in this review are derived from previously published studies and publicly available sources, which are cited within the article.
Conceptualization, SPA and IUO; methodology, SPA; software, SPA; validation, CNE, IUO and MJO; formal analysis, SPA; investigation, IUO; resources, SPA; writing—original draft preparation, SPA; writing—review and editing, SPA and MJO; visualization, SPA and IUO; supervision, CNE and IUO. All authors have read and agreed to the published version of the manuscript.
The authors declare that they have no conflicts of interest.
No external funding was received for this study.
The authors gratefully acknowledge the National Root Crops Research Institute (NRCRI), Umudike, Nigeria, for providing an enabling institutional environment and access to literature, resources, and discussions that supported the preparation of this review. We also appreciate the constructive input and guidance of NRCRI scientific and technical staff, whose expertise and professional advice contributed to shaping the manuscript.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
Abah SP, Okpani MJ, Okwuonu IU, Egesi CN. Genomics-Assisted Cassava Breeding: A Comprehensive Review of Advances, Tools, and Future Prospects. Crop Breed Genet Genom. 2026;8(1):e260002. https://doi.org/10.20900/cbgg20260002.

Copyright © Hapres Co., Ltd. Privacy Policy | Terms and Conditions




