Location: Home >> Detail
Crop Breed Genet Genom. 2026;8(2):e260016. https://doi.org/10.20900/cbgg20260016
 ,
Manje Gowda 1 ,
Leocadio Martinez 2 ,
Juan Burgueno 2 ,
Yoseph Beyene 1 ,
Prasanna M. Boddupalli 1,3
,
Manje Gowda 1 ,
Leocadio Martinez 2 ,
Juan Burgueno 2 ,
Yoseph Beyene 1 ,
Prasanna M. Boddupalli 1,3
1
2
3
*
Natural In vivo maternal haploid induction is a unique and valuable trait in maize, enabling rapid development of completely homozygous inbred lines. This process is facilitated by specialized pollen parents, known as maternal haploid inducers, which induce the formation of seeds containing haploid embryos alongside normal triploid endosperms. The haploid induction rate (HIR) can vary significantly depending on the inducer genotype, source germplasm, and environmental conditions during induction crosses. We conducted a diallel mating among 13 tropical maize inbred lines—five with high inducibility (CML533, CML254, CML451, CML376, CML383), four with moderate inducibility (CML381, CML442, CML435, CML484), and four with low inducibility (CML510, CML396, CML398, CML364)—to study the genetic control of HIR. Ninety-five F1 hybrids, along with their 13 parental lines, were crossed with a common haploid inducer in two different environments, and each set was evaluated for HIR. HIR was assessed in the field based on morphological differences between haploids and diploids, such as plant vigor and leaf erectness. Results revealed substantial variation in HIR among the lines and hybrids. Both general combining ability (GCA) and specific combining ability (SCA) effects were significant for HIR, with five lines showing significant positive GCA effects. Additive genetic effects were predominant, indicating that source germplasm responsiveness to haploid induction can be effectively improved through selection. GCA of parental lines can be effectively used to predict the performance of the hybrids for haploid induction with high accuracy. These findings provide valuable insights for enhancing haploid induction efficiency and accelerating doubled haploid line production in tropical maize breeding programs.
DH, doubled haploid; HI, haploid induction; HIR, haploid induction rate; GCA, general combining ability; SCA, specific combining ability; QTL, quantitative trait loci; GWAS, genome wide association study
The use of haploids in hybrid maize breeding has become a standard practice for generating completely homozygous inbred lines. Doubled haploid (DH) lines derived from haploids offer multiple advantages over conventional inbred lines, including increased genetic gain per cycle, accelerated hybrid development, improved efficiency of molecular marker applications, and streamlined breeding logistics [1,2]. These advantages collectively enhance the overall efficiency of maize breeding programs.
In many crop species, haploid production relies on in vitro techniques such as anther culture or pollen culture, which require specialized laboratory facilities and skilled personnel [3]. However, maize uniquely employs in vivo haploid induction, allowing for the easy and cost-effective generation of haploids in the field [4]. This process depends on rare maize genotypes exhibiting reproductive anomalies that enable haploid induction [5–9]. These genotypes, termed haploid inducers, can be classified into maternal and paternal haploid inducers based on the genetic origin of the induced haploids [10]. Maternal haploid inducers are preferred for large-scale DH line production due to their high haploid induction efficiency [1]. When maternal haploid inducer pollen is used to fertilize maize populations, a proportion of the resulting seeds develop haploid embryos carrying only the maternal genome. The capability of haploid inducers to induce haploids is quantified as the haploid induction rate (HIR), which varies based on the inducer genotype, environmental conditions, and the genetic background of the source germplasm.
The effect of inducer genotypes on HIR is well characterized. The first reported maternal haploid inducer exhibited an average HIR of 1–3% [6], while modern inducers now achieve HIRs of >10% [11,12], enabling large-scale DH production. The genetic basis of the maternal haploid induction trait was well researched, and several quantitative trait loci (QTLs) affecting the HIR were identified [13–18]. Notably, a major effect functional polymorphism in a sperm specific phospholipase gene (zmMTL/NLD/PLA1) was identified as a key determinant of haploid induction [19–21]. Additionally, a mutation in the ZmDMP gene was found to enhance haploid induction in combination with the ZmMTL mutation [22]. Beyond these major loci, several minor QTLs also contribute to variation in HIR [18].
The influence of the environment on HIR was also noted in previous studies, which reported higher HIRs in favorable environments compared to stressful conditions. For instance, Mexican winter seasons with optimal temperatures yielded higher HIRs than hot summer conditions [23]. Additionally, greater environmental effects on HIR were noted in temperate germplasm [2,24]. In contrast to these observations, other studies found no significant environmental effects on HIR [25,26].
Beyond the inducer genotype and environment, the source germplasm also significantly impacts HIR. The effect of maternal genotype on HIR was first observed by Chase in 1949 [27] and subsequently confirmed by multiple studies [2,23,26,28–30]. A detailed analysis of HIR in 671 tropical inbred lines indicated very high genetic variance for HIR, and the genome-wide association study (GWAS) identified several QTLs conditioning the response of the source germplasm to haploid induction [30]. Few studies also explored the combining ability of inbred lines for HIR. Kebede et al. (2011) [23] conducted a half-diallel analysis using 10 tropical inbred lines and found significant general combining ability (GCA) effects, but no significant effects for specific combining ability (SCA) or their interactions with environment. Conversely, De La Fuente et al. (2018) [24] observed significant GCA, SCA, reciprocal, environmental, and interaction effects in a half-diallel study involving three high-HIR and three low-HIR inbred lines.
Efficient and cost-effective production of DH lines depends on producing enough haploid kernels. However, some tropical germplasm—including elite adapted lines and populations derived from them—shows low levels of haploid induction. This limits the success in obtaining enough number of DH lines from such germplasm. To better understand and address this limitation, we previously evaluated a large set of 671 tropical inbred lines for their haploid inducibility and observed substantial variation in HIR among the lines [30]. Based on these results, we selected four inbred lines with low HIR (3–7%), four with moderate HIR (8–11%), and five with high HIR (12–21%) for further investigation. The current study employs a diallel mating design involving these 13 tropical inbred lines to examine the inheritance patterns and combining ability of maternal effects on haploid induction. Additionally, the study aims to assess the predictability of hybrid inducibility based on mid-parent values and GCA effects.
In our earlier study, 671 elite inbred lines adapted to the tropics and subtropics in a wide range of environments, including Latin America, sub-Saharan Africa, and Asia, were characterized for their response to HIR, which indicated a wider variation for HIR in these selected tropical inbred lines [30]. Based on the HIR, a total of thirteen CIMMYT maize lines (CMLs) were selected and crossed in a half diallelic manner. The parents in the diallel experiment included four CMLs with low HIR, four CMLs with medium HIR, and five CMLs with high HIR (Table 1). Pedigrees, adaptation of the inbred lines, and the country where these inbreds were developed are indicated in Table 1. Seeds for these inbred lines were obtained from the CIMMYT maize germplasm bank. A set of 95 F1 crosses were successfully made in the winter cycle of 2018 at the CIMMYT experimental station at Agua Fria (20.26°N, 97.38°W) in Mexico, while a few crosses could not be produced due to non-synchrony for flowering among the inbred lines.
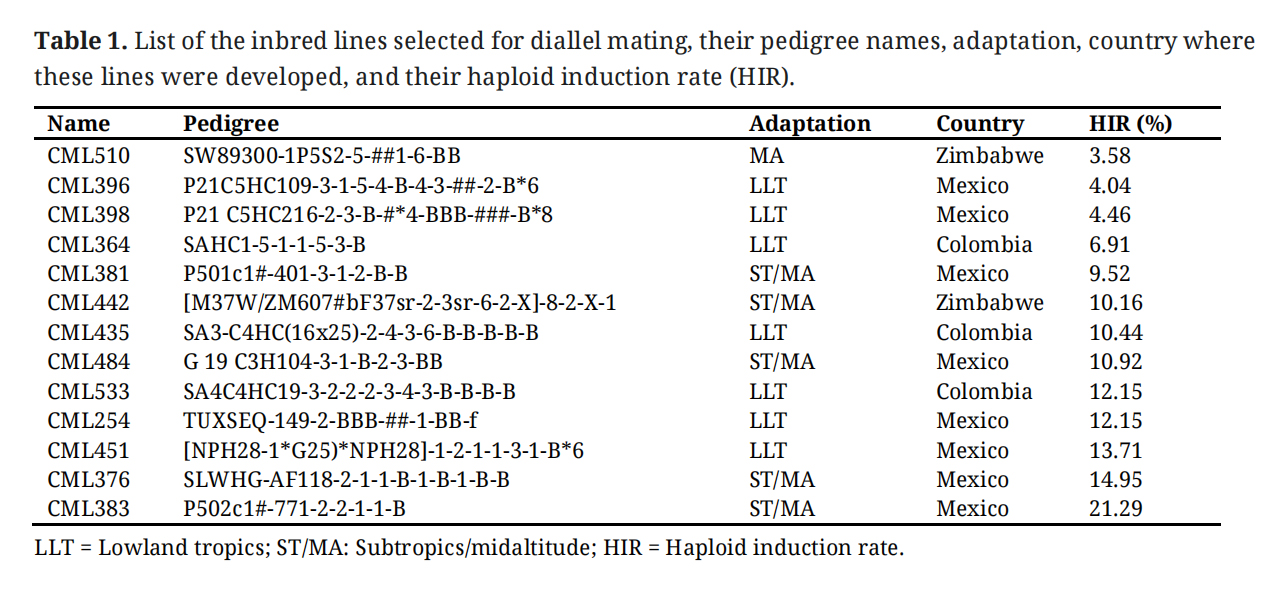 Table 1.
List of the inbred lines selected for diallel mating, their pedigree names, adaptation, country where these lines were developed, and their haploid induction rate (HIR).
Table 1.
List of the inbred lines selected for diallel mating, their pedigree names, adaptation, country where these lines were developed, and their haploid induction rate (HIR).
The di-allelic hybrids, along with 13 parental lines, were crossed to the CIMMYT’s tropical haploid inducer hybrid CIM2GTAIL009xCIM2GTAIL006 [5] in the 2018 summer cycle at the Agua Fria station to induce haploids. The induction crosses were analyzed for HIR by planting the induced seed in the 2019 winter season at Agua Fria in Mexico. The genotypes were crossed to the tropical haploid inducer hybrid CIM2GTAIL009 × CIM2GTAIL006 again in the 2019 summer cycle at Agua Fria station to induce haploids. The induction crosses were analyzed for HIR by planting the induced seed in the winter cycle of 2020 at Metztitlan (20.6°N, 98.76°W) in Mexico.
For assessing HIR, seeds resulting from induction crosses were planted in the field in an alpha lattice design with two replications and 18 blocks with six genotypes per block. For each genotype, 200 seeds were planted. The induced seeds were planted in ridges at a spacing of 75 cm between ridges and 10 cm between plants to accommodate a large number of plants. Haploids and diploids were distinguished based on plant vigor, erectness, and paleness of leaves and purple stem color after three weeks of planting [31]. Diploid plants were removed afterwards, but any doubtful plants were left till flowering, by which the ploidy can be established accurately. HIR was calculated as [(number of true haploids/numbers of surviving plants) × 100] [30]. Non-germinated seeds and plants that died before HIR evaluation were not considered in HIR determination.
GenotypingDNA from all 13 parental inbred lines was extracted and genotyped using a Genotyping by Sequencing (GBS) platform at the Institute for Genomic Diversity, Cornell University, Ithaca, USA, as per the procedure described in earlier studies [30,32]. Quality control on raw GBS SNPs, where a minor allele frequency < 0.05, heterozygosity of >5% and missing data rates > 15% were removed by using TASSEL ver 5.2 [33] and selected 109,590 SNPs were used for further analysis. The genetic relationship among the lines was determined based on the neighbor joining tree algorithm using the phylogenetic tree analysis in TASSEL software v5.2.93. The principal component analysis (PCA) was conducted using the TASSEL software and then visualized by using R software (http://www.R-project.org/).
Statistical AnalysisThe trait HIR data is based on a percentage, so it was checked for statistical model fitting, to know whether it follows normal distribution, has constant variance, and independence or not [34]. Plotting residuals against fitted values showed that the residuals were symmetrically distributed with constant variance; thus, the data were not transformed. Phenotypic data analyses were carried out using Multi-Environment Trait Analysis software in the R environment (META-R) [35,36]. Best linear unbiased predictions (BLUPs) and best linear unbiased estimates (BLUEs) for each parental line and hybrids were generated (Table S1). Analysis of variance was determined for HIR for inbred lines and hybrids by restricted maximum likelihood method using ASReml-R [37]. Dummy variables were used to separate genotypes into parental lines and hybrids. The phenotypic data of the parental inbred lines and hybrids were analyzed based on following linear model:
Further, for the diallel analyses, the software ASReml-R [37] was used. Parental lines GCA and SCA effects of F1 hybrids, as well as their variance components, were computed across environments, using the following linear mixed model (2):
Variance components for GCA of male and female parents, specific combining ability (SCA; pedigree), and their interactions with environments were estimated using a mixed-effects model. Environment and replication nested within environment were fitted as fixed effects, whereas male, female, pedigree, and their interactions with environment were treated as random effects, assuming independent normal distributions with a mean of zero. The kinship matrix was estimated by using the VanRaden algorithm [38]. For male and female effects, we considered a variance-covariance matrix based on the van Raden genomic relationship which we obtained from markers data.
To get the line effect without considering its role as male or female (both) we used the “equate.levels” instruction of ASReml. The significance of GCA and SCA effects were tested with a z-test, using standard errors of GCA and SCA effects, respectively. The variance components of GCA (σ2GCA) and SCA (σ2SCA) were estimated from the corresponding combining ability effects. The baker’s ratio [39] was used to evaluate the relative importance of GCA and SCA. The ratio of phenotypic variance contributed by the genetic variance was used to compute broad-sense heritability based on the entry means [40]. The ratio of GCA effects (2GCA) to total genetic effects (2GCA + SCA) closer to unity is considered a high predictability of HIR performance. Pearson’s correlation coefficients (r) were calculated between the F1 hybrids and mid-parent values. A leave-one-hybrid-out cross-validation procedure described by Schrag et al. (2006) [41] was implemented to determine the coefficients of the correlations between F1 hybrids and the sum of GCA effects of both parents.
The analysis of HIR among the inbred lines used in this study revealed significant variability among the lines (Table 1 and Figure 1A). The lines CML510, CML396, CML398, and CML364 showed low HIR ranging from 3.58 to 6.91, whereas the lines CML381, CML442, CML435, and CML484 showed medium levels of HIR (9.52–10.92%). The lines CML533, CML254, CML451, CML376, and CML383 showed higher HIR (12.15% to 21.29%). The HIR values of the parental lines and F1 hybrids followed the normal distribution pattern (Figure 1). The HIR was ranged from 2% to 21% for parental lines and 9% to 17% for hybrids as shown in Figure 1.
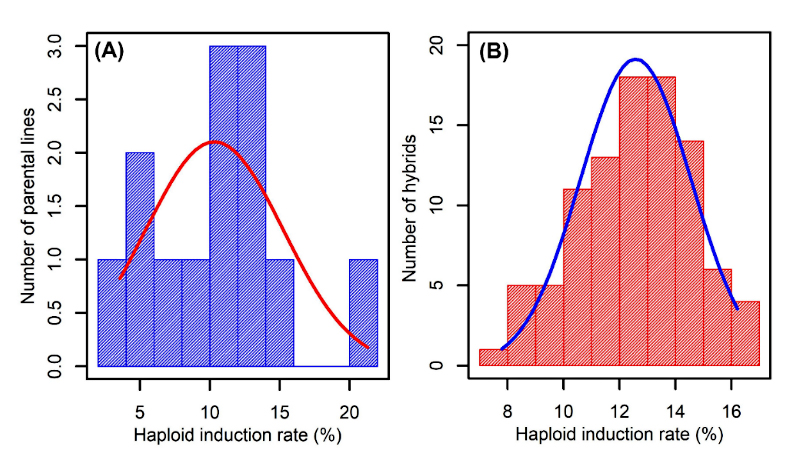 Figure 1.
Histogram and normal distribution profile of HIR for parental lines (A) and hybrids (B).
Figure 1.
Histogram and normal distribution profile of HIR for parental lines (A) and hybrids (B).
Among the thirteen inbred lines used in this study, seven were developed for lowland tropical regions, and the remaining six inbreds were developed for midaltitude/subtropical regions (Table 1). Based on GBS SNPs data, the lowland tropical inbreds and mid-altitude/subtropical inbreds fell into two distinct clusters (Figure 2A). Among the lowland tropical inbreds, CML533, CML435, and CML364 inbreds that were developed by the CIMMYT breeding program in Colombia formed a separate cluster. Another set of lowland inbreds CML396, CML398, CML254, and CML451, that were developed by CIMMYT in Mexico, clustered together. Subtropical inbreds CML376 and CML383, together with CML381, were developed by the subtropical breeding program in Mexico are clustered together. The subtropical inbreds CML442 and CML510, developed in Zimbabwe together with CML484, developed in Mexico formed a separate cluster. The PCA analysis revealed a significant variability among the inbred lines (Figure 2B). The first two principal components accounted for 27% of the total variation.
To evaluate the potential impact of population structure on GCA estimates, we analyzed Model 2 including the first two principal components (PC1 and PC2) from the PCA analysis as fixed effects. Neither PC was significant (Wald test: p = 0.0700 for PC1 and p = 0.3816 for PC2), indicating that population structure did not contribute significantly to variation in HIR. Furthermore, the estimated variance components were virtually unchanged compared with the original model (GCAmale = 1.08, GCAfemale = 3.19, GCAmale × Environment = 0, GCAfemale × Environment = 0.18, SCA = 1.04, SCA × Environment = 2.72, Residual = 7.59). These results demonstrate that the observed GCA effects are robust to adjustment for population structure and are not inflated by stratification among the Colombian, Mexican, and Zimbabwean breeding groups identified by PCA (Figure 2B).
The diallelic crosses were grouped into six categories based on the haploid induction rate (HIR) of the parental lines (Table 2, Figure 3, Table S1). Hybrids made between two high-HIR lines had an average HIR of 14.5%, while those between high- and medium-HIR lines averaged 14.0% of HIR. In F1 hybrids with both parents having high HIR, the HIR ranged from 12.84% to 15.60%. Crosses between one high-HIR parent and one medium-HIR parent, the HIR ranged from 11.95% to 15.90%. F1 hybrids developed by using medium × medium HIR parents averaged 13.24% of HIR. Hybrids involving low-HIR parents (high × low, medium × low, low × low) all had average HIR below 12%, with low × low HIR crosses showing the lowest average HIR (8.35%) (Figure 3). All low × low crosses had less than 10% HIR, except for F1 hybrids developed from CML364 × CML510.
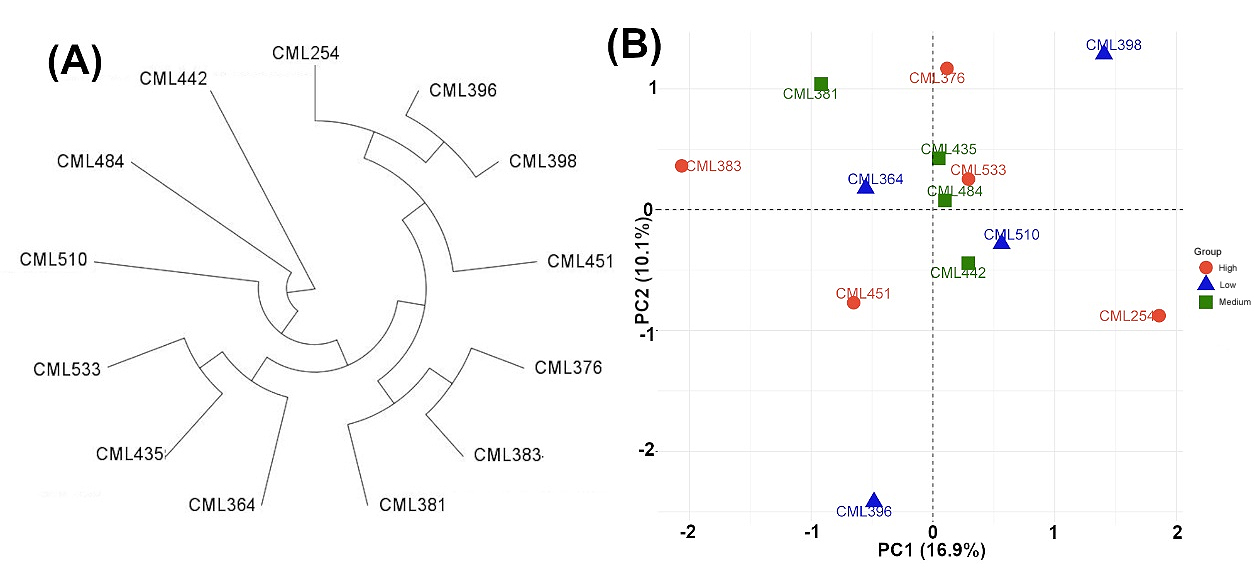 Figure 2.
Dendrogram (A) and principal component analysis (B) of 13 CMLs based on the neighbor-joining method estimated using 109,590 SNP markers.
Figure 2.
Dendrogram (A) and principal component analysis (B) of 13 CMLs based on the neighbor-joining method estimated using 109,590 SNP markers.
 Table 2. Mean performance of F1 hybrids across two environments for HIR in the diallel experiment.
Table 2. Mean performance of F1 hybrids across two environments for HIR in the diallel experiment.
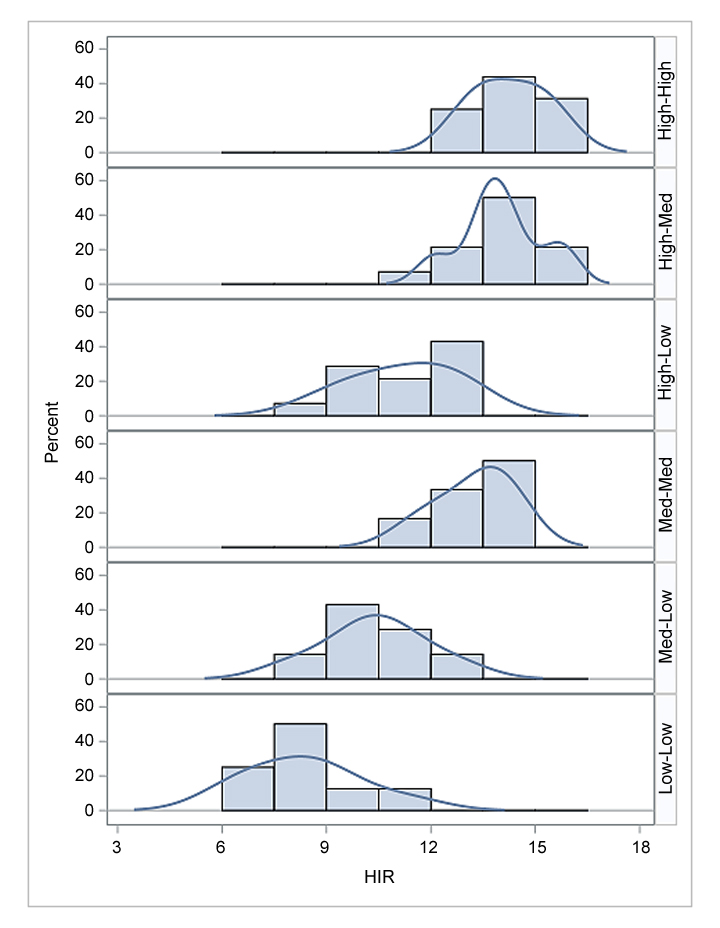 Figure 3.
Histogram and kernel density of hybrids for haploid induction rate (HIR %) across two environments in diallel experiment.
Figure 3.
Histogram and kernel density of hybrids for haploid induction rate (HIR %) across two environments in diallel experiment.
Analyses of variance (ANOVA) indicated significant genotypic variation for both parental lines and F1 hybrids for HIR (Table 3). Hybrids tend to show slightly higher HIR compared to the average HIR of parental lines. Genotype × environment interaction variance is also significant for both parental lines and F1 hybrids. The inbreds showed high heritability (0.67) for HIR, similar to hybrids which showed a heritability of 0.68. When variance components are further partitioned into GCA and SCA (with Model 2), assuming that male and female parents are not contributing equally, the GCA for both male and female parents in the diallelic crosses is significant (Table 4). GCA for female parents and its interaction with the environment, GCA for male × female × environment is also significant (Table 4). SCA is less significant compared to GCA; however, SCA × Env is highly significant. Additive variance is 4.4 times the dominance variance. Broad-sense heritability is higher than narrow-sense heritability. When the male and female parents are assumed to be equal contributors, variance for both lines and hybrids was significant (Table 4). However, the parental line x environment interaction was not significant, but the hybrid × environment interaction was significant.
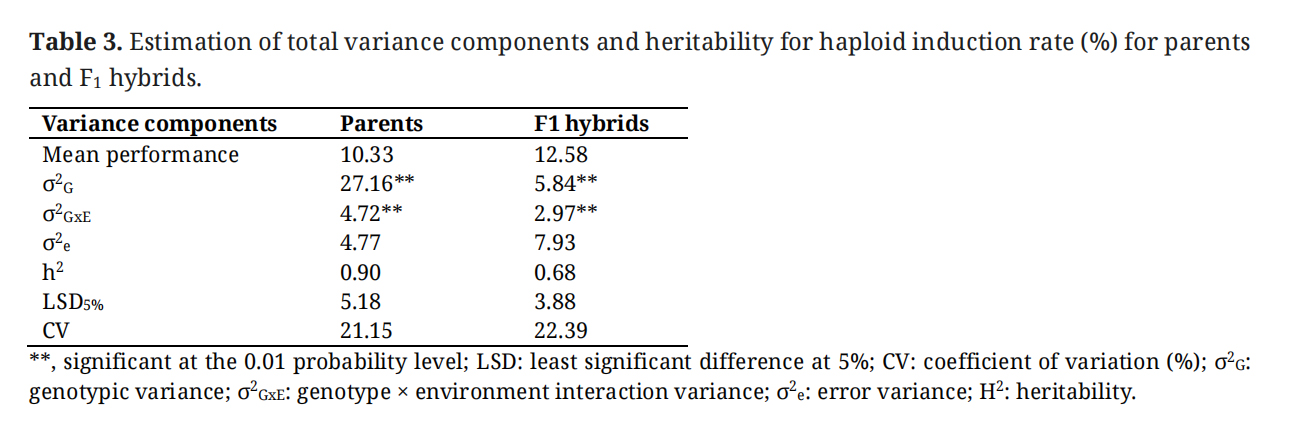 Table 3.
Estimation of total variance components and heritability for haploid induction rate (%) for parents and F1 hybrids.
Table 3.
Estimation of total variance components and heritability for haploid induction rate (%) for parents and F1 hybrids.
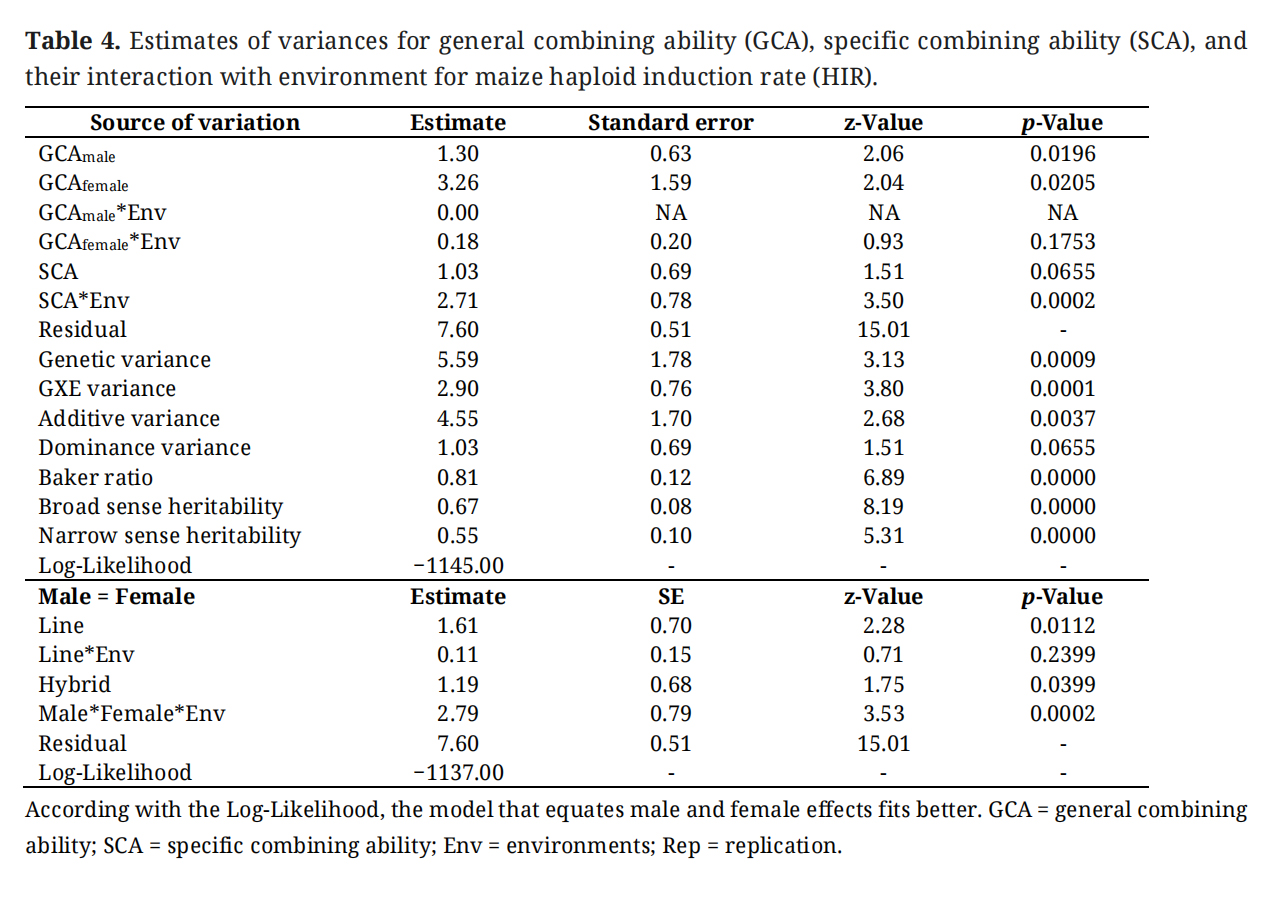 Table 4.
Estimates of variances for general combining ability (GCA), specific combining ability (SCA), and their interaction with environment for maize haploid induction rate (HIR).
Table 4.
Estimates of variances for general combining ability (GCA), specific combining ability (SCA), and their interaction with environment for maize haploid induction rate (HIR).
GCA for inbreds with low HIR tends to be low, contributing negatively whether used as a male or female parent in the diallelic crosses. The lines CML396 and CM398 had the highest negative GCA effects for HIR. GCA of the inbreds with high HIR tends to be positive when used as either male or female parents (Table 5). The lines CML383 and CML376 had the highest positive GCA effects among all the inbreds included in the study. GCA effects for the inbreds with medium HIR are mostly in a positive direction except for CML381 when used as a female parent and CML442 when used as a male parent. SCA effects for each cross combination were estimated based on phenotypic and genotypic data (Table 6). SCA variance was not significant (p = 0.06), and no individual effect was statistically different from zero minimum (p = 0.0601). The SCA effects ranged from −1.29% to 1.67% with and standard error of 0.89%. Mid-parent performance was significantly (p < 0.01) correlated with F1 hybrid performance for HIR (r = 0.76, Figure 4). However, compared to predictions based on mid parent values, the GCA based correlations for F1 hybrid performance was higher with r = 0.96 (p < 0.01) (Figure 4).
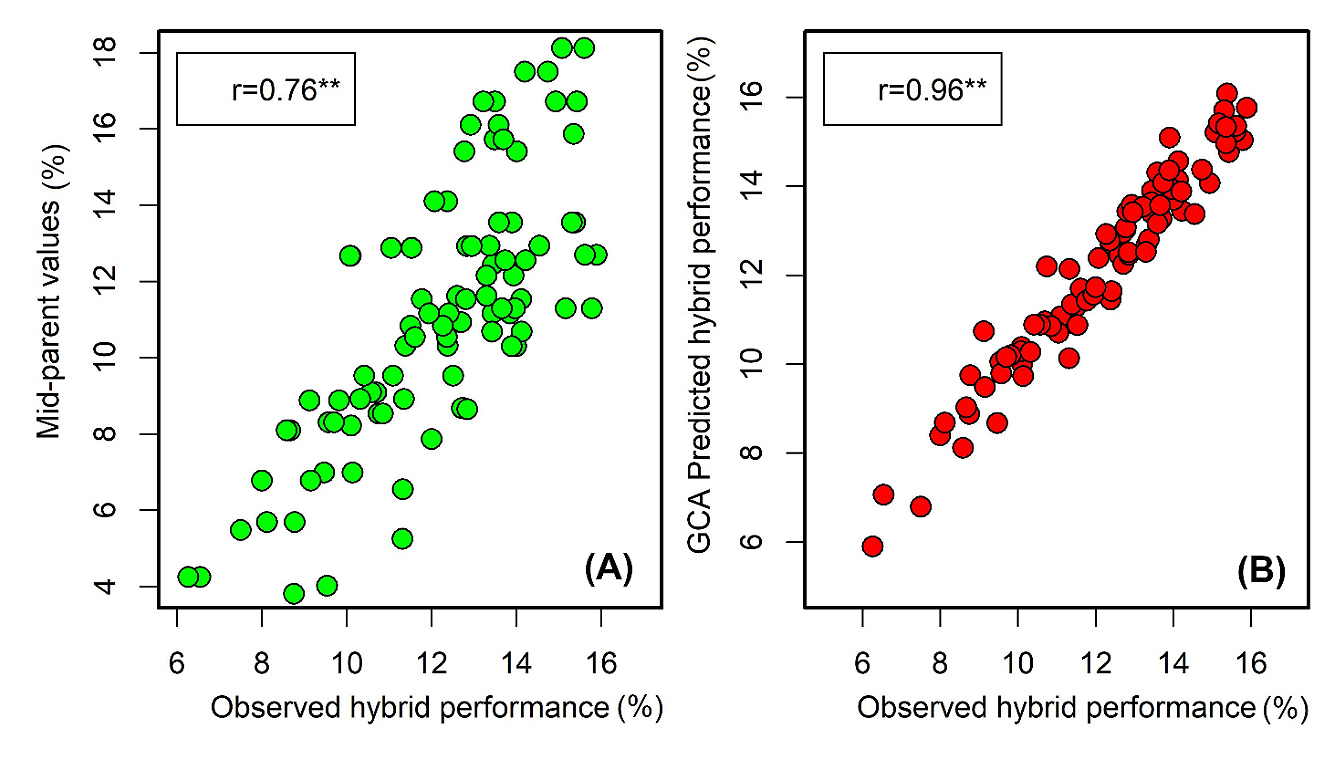 Figure 4.
Association of (A) mid parent values and (B) GCA based prediction with observed F1 hybrid performance for haploid induction rate (%). ** Means a significant correlation with p-value < 0.01.
Figure 4.
Association of (A) mid parent values and (B) GCA based prediction with observed F1 hybrid performance for haploid induction rate (%). ** Means a significant correlation with p-value < 0.01.
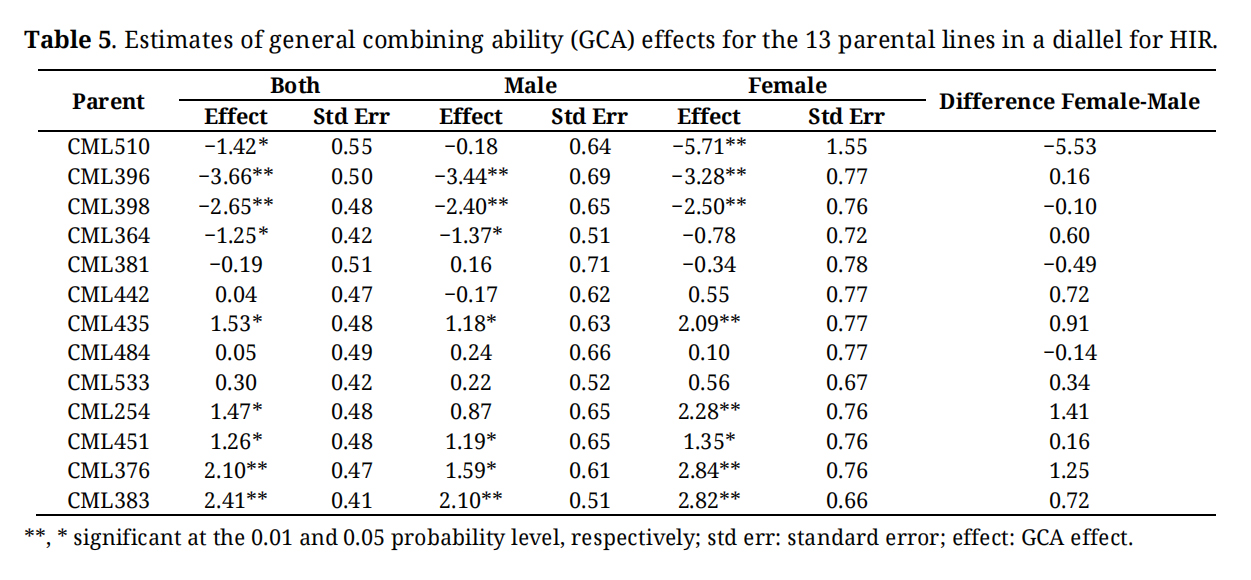 Table 5. Estimates of general combining ability (GCA) effects for the 13 parental lines in a diallel for HIR.
Table 5. Estimates of general combining ability (GCA) effects for the 13 parental lines in a diallel for HIR.
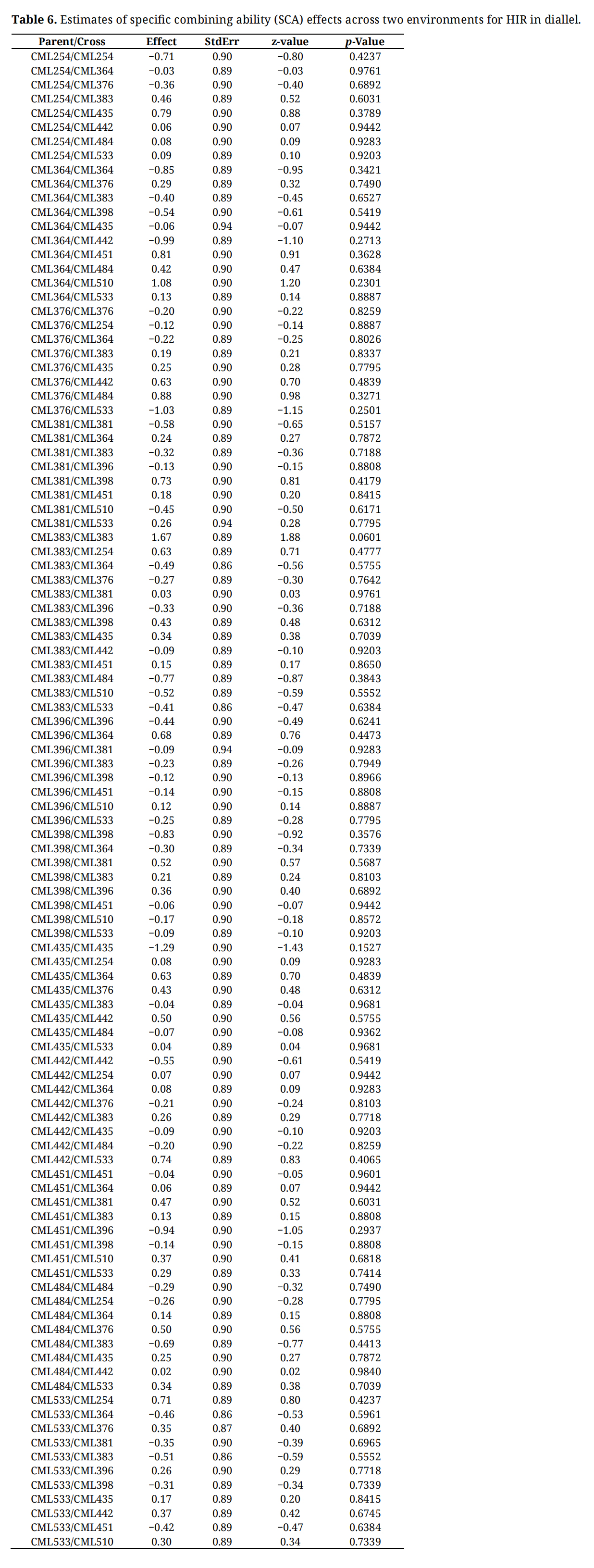 Table 6. Estimates of specific combining ability (SCA) effects across two environments for HIR in diallel.
Table 6. Estimates of specific combining ability (SCA) effects across two environments for HIR in diallel.
Producing haploids in desired numbers economically is an important requirement in the maize DH line production process. Maternal haploid inducer genotypes with an innate ability to produce haploids are central to this process. Improvements in HIR in maternal haploid inducers were achieved through decades of selection for HIR, with modern maternal haploid inducers having HIR > 10% compared to the first reported inducer with ~1% of HIR [1,16]. While HIR is relatively stable when assessed within a given inducer genotype and environment, it varies considerably when the same inducer is used across different female genotypes. This highlights an important opportunity to improve not only the innate HIR of inducers but also the genotypic response of source germplasm to haploid induction.
Some female genotypes consistently show higher HIR responses, suggesting the presence of genetic factors within such source germplasm influencing haploid induction efficiency [30]. Understanding these genotypic responses has practical implications. Knowledge of germplasm performance for haploid induction allows breeders to plan more precisely, optimizing field space, labor, and other inputs during the haploid induction stage. Identification and incorporation of rare, high-responding genotypes into breeding pipelines is strategically beneficial in enhancing the efficiency and cost-effectiveness of DH line production in the long term.
Estimating variance components and combining ability for HIR response could provide valuable insights into the inheritance of HIR, with significant general and specific combining ability effects guiding parental selection to maximize haploid induction efficiency. In this diallelic study, tropical inbred lines adapted to the lowland tropics and subtropics exhibited considerable variation for HIR, ranging from 3.58% to 21.29%, reaffirming that the source germplasm from which haploids are derived plays a significant role in determining HIR. The thirteen inbreds evaluated here were previously characterized for their HIR using a different haploid inducer [30], and the HIR trends observed in this study were consistent with the earlier findings. However, all inbreds showed higher HIR levels in the present study, which can be attributed to the use of a different haploid inducer with enhanced haploid induction capability [5] compared to the inducers used in the earlier study [30]. Similar to inbred lines, the hybrids also showed great variation for HIR, ranging from 5.5% to 17.99% (Table S1). This variability among both the parental lines and hybrids for HIR implies strong potential for improving haploid induction response through selection in source germplasm.
There are several indications to confirm a strong influence of source germplasm on HIR, alongside variation for HIR observed among the inbreds and hybrids. First, there is a significant genotype effect observed in ANOVA for both the lines and the F1 hybrids (Tables 2 and 3). Secondly, the hybrids derived from high HIR inbreds consistently exhibited high HIR, while those from low HIR parents showed lower HIR. Thirdly, a large broad-sense heritability (h2 = 0.67) suggests that most of the phenotypic variation observed was attributable to genetic differences in the germplasm evaluated. This conclusion is in agreement with observations from several other studies on HIR using temperate and tropical maize germplasm [2,23,26–30].
The total variance for genotype x environment interaction is significant for both parental inbreds and F1 hybrids, highlighting the critical role of environmental factors in HIR expression and the broader adaptation of the material studied. Previous studies also observed a significant effect of environment on HIR in both temperate [2,24] and tropical maize germplasm [23], where higher HIRs were generally achieved under optimal environmental conditions. In contrast to these observations, other studies found no significant environmental effects on HIR [25,26]. In our study, GCA × environment interaction variances were nonsignificant, indicating that parental inbreds performed consistently in hybrid combinations across environments, consistent with Kebede et al. (2011) [23] but differing from De La Fuente et al. (2018) [24]. The significant SCA × environment-interaction variance observed here suggests that hybrid performance for HIR is not solely determined by the average effects of the parents, but also by how specific parental combinations interact with particular environmental conditions.
Our results are broadly consistent with those of De La Fuente et al. (2018) [24], who reported significant GCA, SCA, environmental, and GCA × environment effects for haploid inducibility. However, the magnitude of GCA effects observed in the present tropical germplasm was considerably greater, with a maximum GCA of 2.41% compared with 0.06% reported by De La Fuente et al. (2018) [24] and 1.06% by Kebede et al. (2011) [23]. This likely reflects the wider range of parental HIR (3.58–21.29%) and the broader genetic diversity represented by CIMMYT tropical germplasm, encompassing distinct lowland tropical, subtropical, and mid-altitude breeding pools. Significant GCA and SCA variances indicate that both additive and non-additive genetic effects contribute to HIR, although the predominance of additive gene action was evident from the larger GCA variance, high Baker’s ratio (0.81), greater additive than dominance variance, and medium narrow-sense heritability (h2 = 0.55). Collectively, these results demonstrate that responsiveness to haploid induction is largely under additive genetic control, enabling effective improvement through selection and increasing the predictability of hybrid performance from parental breeding values.
Since GCA effects were more predominant than SCA effects in governing HIR, breeding strategies to improve HIR should primarily exploit additive genetic variation. The inbred lines CML383, CML376, CML254, CML451, and CML435 exhibited the highest positive GCA effects, indicating their ability to consistently transmit favorable alleles for haploid inducibility to progeny. These lines represent valuable donor parents for enhancing HIR and can be integrated into CIMMYT’s forward breeding pipeline through recurrent crossing and DH line development, similar to the deployment of favorable alleles for MLN and MSV resistance. The associated QTLs can be targeted through marker-assisted or genomic selection to accelerate the accumulation of favorable alleles while maintaining gains for agronomic performance and disease resistance, ultimately improving induction efficiency, reducing DH production costs, and increasing genetic gain.
Significant SCA × environment interactions observed in this study indicate that specific parental combinations responded differently across environments, suggesting that non-additive genetic effects underlying inducibility are sensitive to environmental conditions. One plausible explanation is variation in temperature and related stresses during pollination and fertilization, which influence pollen viability, fertilization success, embryo development, and genome elimination processes associated with haploid induction. Earlier studies [23,24] reported reduced HIR under hot summer conditions compared with more favorable environments, indicating that environmental stress can affect induction efficiency. Tropical haploid inducers may also differ in their adaptation to high-temperature environments, which could contribute to differential hybrid responses and the observed SCA × environment interactions. Further studies evaluating induction crosses under controlled temperature regimes would help clarify the physiological mechanisms responsible for these genotype-specific environmental responses.
Parental lines for hybrid development are often selected based on their per se performance. For simply inherited traits, mid-parent values can predict hybrid performance with moderate to high accuracy (>0.60), as reported for days to silking, ear dry matter content, and plant height in maize [42] and for plant height, heading time, and 1000-kernel weight in wheat and triticale [43]. In this study, the correlation between mean parental values and F₁ performance for HIR was moderately high, indicating that mid-parent values can guide preliminary hybrid selection, although perfect prediction is not assured.
For traits predominantly governed by additive variance (GCA), prediction accuracy is generally higher than for those with substantial non-additive effects (SCA) as shown for dry matter content in maize [42]. GCA in the present study was a strong predictor of hybrid HIR, achieving a prediction accuracy of 0.96 (p < 0.01) (Figure 4), like the previous results for maize lethal necrosis resistance [44]. Prior information on GCA values for the target trait can therefore greatly assist breeders in identifying lines with the highest probability of producing high HIR resulting in production of more DH lines with less resources. Overall, these findings suggest that for HIR, GCA alone can serve as a robust predictor for identifying optimal parental combinations to produce DH lines with high haploid induction rates. A comprehensive understanding of the trait’s genetic architecture—including variance components, heritability, combining ability, and trait-linked markers—further improves the precision of germplasm selection, ensuring more efficient and targeted DH-based breeding pipelines.
In conclusion, this study confirms that HIR in tropical maize is strongly influenced by the genetic background of the source germplasm. The predominance of additive genetic effects, evidenced by high GCA variances, Baker’s ratio, and narrow-sense heritability, underscores the potential for substantial genetic gains to improve HIR through selection of more responsive germplasm. Lines such as CML383, CML376, CML254, CML451, and CML435 emerged as valuable donors of favorable alleles for HIR enhancement. The strong correlation between mid-parent values and hybrid performance, coupled with high prediction accuracy from GCA, indicates that breeders can reliably use per se performance and GCA estimates to identify optimal germplasm for DH production with optimal resources. By integrating these insights, breeding programs can systematically improve haploid induction efficiency, reduce DH production costs, and accelerate genetic gains in maize improvement pipelines.
The following supplementary materials are available online, Table S1: BLUEs and BLUPs of 13 parental lines and F1 hybrids evaluated two locations for haploid induction rate.
The dataset of the study is available from the authors upon reasonable request.
Conceptualization, funding acquisition, project & resources administration, PBM and VC; methodology, investigations, formal analysis and visualization, VC, MG, YB, JB, and LM; supervision, VC, MG, PBM. and YB; Original draft preparation, VC and MG; writing-review and editing; all authors.
Authors declare that they have no known competing personal or financial interests that could have appeared to influence the results of this study.
The research was supported by the Bill and Melinda Gates Foundation (B&MGF), and the United States Agency for International Development (USAID) through the Stress Tolerant Maize for Africa (STMA, B&MGF Grant # OPP1134248) Project, AG2MW (Accelerating Genetic Gains in Maize and Wheat for Improved Livelihoods, B&MGF Investment ID INV-003439) project.
The authors are grateful to the International Maize and Wheat Improvement Center (CIMMYT) scientists and technicians who generated the germplasm, and highly appreciate the technical support received from the staff members affiliated to CIMMYT maize research station in Agua frea, Mexico.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
Chaikam V, Gowda M, Martinez L, Burgueno J, Beyene Y, Prasanna BM. Partial Diallelic Analysis of Maternal Effects on In Vivo Haploid Induction in Tropical Maize Germplasm. Crop Breed Genet Genom. 2026;8(2):e260016. https://doi.org/10.20900/cbgg20260016.

Copyright © Hapres Co., Ltd. Privacy Policy | Terms and Conditions




